Introduction au contrôle de la corrosion, du tartre et de l’encrassement biologique dans les systèmes de refroidissement
Le chapitre précédent a décrit les principes fondamentaux de la conception et du fonctionnement de la tour de refroidissement et de l’échangeur de chaleur. Dans ce chapitre, nous examinons la chimie de l’eau de refroidissement et les programmes de traitement pour maintenir la fiabilité dans l’ensemble du réseau d’eau de refroidissement.
Les systèmes de refroidissement nécessitent une protection contre la corrosion, l’entartrage et l’encrassement biologique (ou l’encrassement microbiologique) pour maximiser les performances. Une représentation symbolique de ces questions et de leur interdépendance est illustrée ci-dessous.

Le contrôle de la corrosion, du tartre et de l’encrassement biologique doit être envisagé de manière holistique. Un quatrième facteur de plus en plus important est l’impact environnemental potentiel de la chimie du traitement de l’eau, en particulier en ce qui concerne les produits chimiques qui pourraient apparaître dans la décharge de l’usine. Les programmes de traitement qui étaient autrefois courants peuvent ne plus être autorisés ou peuvent être sévèrement restreints en raison des règlements sur le congé.
Bien que les méthodes de traitement aient souvent plusieurs fonctions, un aspect clé est la protection des surfaces métalliques. Dans les sections suivantes, nous passerons en revue les mécanismes de corrosion et les méthodes de contrôle les plus courants.
Table des matières
Un bref examen de la métallurgie du système de refroidissement et de la composition des matériaux
Mécanismes de corrosion des métaux primaires
Types de corrosion
Mécanismes de dépôt
Formation de tartre
Contrôle des dépôts et de la corrosion
Contrôle de la corrosion
Chimie de tournage
Contrôle de la corrosion de l’eau de refroidissement fermée (CCW)
Contrôle de la corrosion par sélection des matériaux et électrochimie
Préoccupations microbiologiques
Contrôle microbiologique
Biocides oxydants et non oxydants
Nettoyage et assainissement des encrassements microbiologiques
Méthodes de contrôle de l’encrassement macroscopique
Méthodes de surveillance microbiologique
Surveillance des biocides oxydants
Surveillance de l’alimentation, des dépôts et de la corrosion des produits chimiques
Nettoyage et passivation du système de refroidissement
Conclusion
Un bref examen de la métallurgie du système de refroidissement et de la composition des matériaux
À titre d’examen rapide, le matériau typique pour la tuyauterie du système de refroidissement et de nombreuses coquilles d’échangeur de chaleur (HX) est l’acier au carbone doux. Les tubes ou plaques HX peuvent être en acier inoxydable, en alliages de cuivre, en titane, en aluminium ou, dans certains cas, en alliages résistants à la corrosion coûteux. Les fixations en acier galvanisé sont souvent présentes dans les tours de refroidissement, tandis que les tours plus petites peuvent être principalement galvanisées. La plupart des grandes tours de refroidissement ont des bassins en béton, et certaines ont encore des composants structurels en bois. Ainsi, le système de refroidissement complet peut comprendre une variété de matériaux, où la connaissance de tous est essentielle pour la sélection de programmes de contrôle de la corrosion fiables.
Mécanismes de corrosion des métaux primaires
La corrosion des métaux est un processus électrochimique dans lequel les métaux dans un état raffiné retournent à leur forme naturelle. Le fer est l’exemple classique. Selon la référence 1 « Les gisements de minerai de fer les plus importants de la Terre se trouvent dans les roches sédimentaires. Ils se sont formés à partir de réactions chimiques qui combinaient le fer et l’oxygène dans les eaux marines et les eaux fraîches. Les deux minéraux les plus importants dans ces dépôts sont l’hématite (Fe2O3) et la magnétite (Fe3O4) des oxydes de fer. Ces minerais métalliques ont été extraits pour produire presque tous les objets en fer et en acier que nous utilisons aujourd’hui. » Les matériaux à base de fer et autres métaux mis en service peuvent être attaqués de diverses façons. Le plus observable est la réaction du fer ou de l’acier doux avec de l’eau et de l’oxygène pour produire de la rouille, mais d’autres mécanismes de corrosion sont courants. Les plus répandus comprennent :
- Corrosion générale ou uniforme
- Piqûres
- Corrosion par gravité
- Corrosion par érosion
- Corrosion influencée microbiologiquement
- Fissuration due à la corrosion sous contrainte, fatigue due à la corrosion et corrosion intergranulaire
- Corrosion galvanique
- lixiviation sélective ou déallocation
Un contrôle approprié de la corrosion est nécessaire pour prolonger la durée de vie de l’équipement, minimiser le transport des produits de corrosion vers d’autres emplacements et parfois pour assurer la sécurité des employés.
Cellule de corrosion
La force motrice des réactions corrosives est le potentiel électrique entre l’accepteur d’électrons (le milieu corrosif) et le donneur d’électrons (le métal). L’exemple suivant illustre le processus fondamental de corrosion. Il s’agit de la simple expérience en laboratoire d’immersion d’une barre de fer dans une solution d’acide chlorhydrique (HCl). Presque immédiatement, des bulles apparaîtront tout le long de la surface submergée de la barre, et dans un délai relativement court, la corrosion deviendra clairement visible. La figure 7.2 résume la chimie.
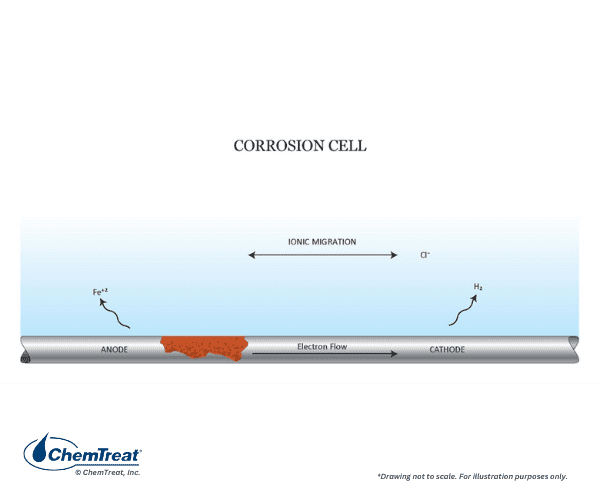
Trois réactions expliquent le processus global :
- Chaque atome de fer sur un site de corrosion abandonne deux électrons (s’oxyde) et passe donc d’un état d’oxydation zéro à un état d’oxydation +2, Fe0 → Fe2+ + 2e– (Éq. 7-1). Les ions Fe2+ (ferreux) migrent dans la solution. Ces sites sont appelés anodes.
- Les électrons libérés dans les anodes circulent dans le métal vers d’autres sites où ils réagissent avec les ions hydrogène (réduction) de l’acide pour produire de l’hydrogène gazeux, 2H+ + 2e– → H2↑ (Éq. 7-2). La réduction se produit au niveau des cathodes.
- Les ions chlorure (Cl–) et les ions ferreux migrent à travers la solution pour produire du chlorure ferreux soluté (FeCl2) et compléter le circuit électrique.
Dans ce cas particulier, des anodes et des cathodes se forment tout le long de la surface métallique. De nombreuses bulles d’hydrogène se forment immédiatement, et peu de temps après, une corrosion visible apparaît sur toute la barre. Il s’agit d’un exemple de corrosion générale, où les anodes et les cathodes se déplacent constamment, et c’est également un exemple classique d’une réaction d’oxydation-réduction ou de « redox ».
Chaque métal a une tendance différente à libérer ou à accepter des électrons, et comme nous le verrons sous peu, l’agent corrosif a une grande influence. La liste des potentiels métalliques des demi-cellules par rapport au potentiel des demi-cellules d’hydrogène est utile et instructive.
2H++ 2e–� sygnema H2↑ E0 = 0,00 V pour une solution molaire par définition
Le tableau suivant présente les potentiels de plusieurs des métaux les plus courants.
Tableau 7-1. Potentiels de demi-cellule pour les métaux bien connus par rapport à la demi-cellule d’hydrogène
| Réaction de l’électrode | Potentiel d’électrode standard (V0) |
| Mg → Mg2+ + 2e– | 2,363 |
| Al → Al3+ + 3e– | 1,662 |
| Zn → Zn2+ + 2e– | 0,763 |
| Fe → Fe2+ + 2e– | 0,440 |
| H2 → 2H+ + 2e– | 0,000 |
| Cu → Cu2+ + 2e– | -0,340 |
| Ag → Ag+ + e– | -0,800 |
| Au → Au3+ + 3e– | -1 420 |
À partir de ce tableau, nous pouvons tirer plusieurs concepts importants.
- Les métaux au-dessus de l’hydrogène dans la table se corroderont tous dans l’acide.
- Considérez à nouveau la réaction illustrée à la Figure 7.2. Le tableau indique que le fer présente un potentiel de réaction significatif par rapport à l’acide.
- Le cuivre (et généralement ses alliages) est du côté « noble » du potentiel de demi-cellule d’hydrogène, ce qui signifie qu’il reste stable dans les acides minéraux communs. Les agents oxydants sont généralement nécessaires pour la corrosion.
- L’argent et l’or sont deux des métaux vraiment nobles. Particulièrement pour l’or, des solutions spéciales, p. ex., aqua regia, sont nécessaires pour dissoudre le métal.
- Ces données sont également importantes pour déterminer la probabilité de corrosion galvanique lorsque deux métaux différents sont couplés dans un environnement d’eau. La corrosion galvanique est examinée plus en détail plus tard.
En ce qui concerne le matériau structurel commun de nombreux systèmes d’eau de refroidissement, la ruelle douce, Figure 7.3 illustre l’influence du pH, c.-à-d. la concentration d’ions d’hydrogène.
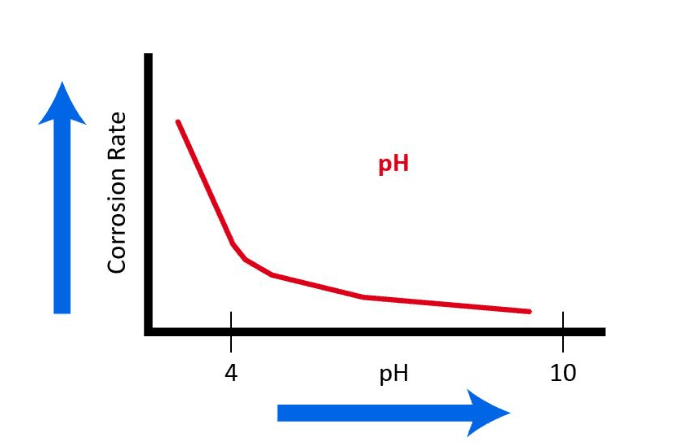
Par conséquent, la plupart des programmes modernes de chimie du système de refroidissement fonctionnent dans une plage de pH supérieure à 7 à 8. Dans ces conditions, une mince couche passive de CaCO3 peut se former pour inhiber davantage la corrosion. Cependant, comme nous en avons brièvement discuté au chapitre 6 et nous le réexaminerons plus tard dans ce chapitre, la formation de tartre de carbonate de calcium peut être l’un des problèmes les plus problématiques dans les systèmes de refroidissement.
Si l’acide était le seul corrodant dans un système d’eau de refroidissement, le contrôle de la corrosion serait simple. Malheureusement, de nombreuses autres réactions cathodiques sont possibles, la plus répandue étant la réduction de l’oxygène dans les solutions neutres ou alcalines.
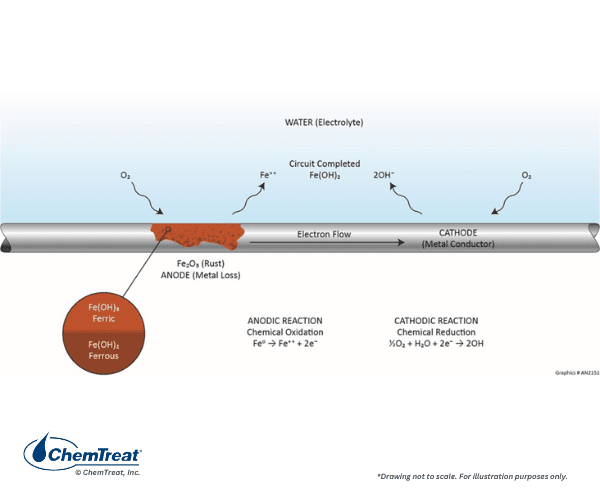
La réaction anodique est la même que celle illustrée précédemment.
- Fe0 → Fe2+ + 2e‒ | Éq. 7-1
L’oxygène est réduit à la cathode :
- ½O2 + H2O + 2e→ 2OH‒ | Éq. 7-3
Les ions d’hydroxyde se combinent avec Fe2+ pour former de l’hydroxyde ferreux :
- Fe2+ + 2OH→ Fe(OH)2 | Éq. 7-4
Dans l’environnement chargé d’oxygène, d’autres réactions se produisent. Tout d’abord, l’hydroxyde ferreux continuera à réagir avec l’oxygène pour former de l’hydroxyde ferrique :
- 2Fe(OH)2 + ½O2 + H2O → 2Fe(OH)3↓ | Éq. 7-5
Les réactifs illustrés dans les équations 7-4 et 7-5 sont en équilibre avec d’autres espèces d’oxyde de fer, comme illustré ci-dessous :
- Fe(OH)2 �. sup. FeO + H2O | Éq. 7-6
- Fe(OH)3 �. sup. FeO(OH) + H2O | Éq. 7-7
Ces produits finissent par se déshydrater en rouille, qui est de couleur brune et n’offre aucune protection au métal sous-jacent :
- 2FeO(OH) �. sup. Fe2O3↓ + H2O | Éq. 7-8
Deux autres aspects importants à prendre en compte sont que dans le processus électrochimique, les réactions cathodiques dictent le taux de corrosion, tandis que les réactions anodiques dictent le type de corrosion. Comme nous le verrons plus en détail, des situations peuvent survenir avec un petit nombre d’anodes fixes dans un grand environnement cathodique. Cette combinaison peut entraîner une corrosion localisée grave et des défaillances potentiellement rapides.
Le lecteur observera que le magnésium et l’aluminium sont au sommet du tableau 7.1, et demandera pourquoi ces matériaux sont utilisés à de nombreuses fins d’infrastructure comme certaines pièces d’avion, certains composants électriques, des canettes de boisson, etc. Les éléments sont tellement réactifs que pendant le processus de production, une couche d’oxyde serrée se forme sur la surface métallique et protège le métal de base de la corrosion. Seuls les acides ou les alcalis forts attaqueront cette couche protectrice, de sorte que les métaux restent assez stables pendant le service normal.
Plusieurs autres facteurs importants influencent la plupart des mécanismes de corrosion. Ceux-ci sont décrits ci-dessous.
Conductivité
La corrosion est un processus électrochimique et, à mesure que la concentration en solides dissous augmente, la conductivité et le taux de corrosion correspondants augmentent également.
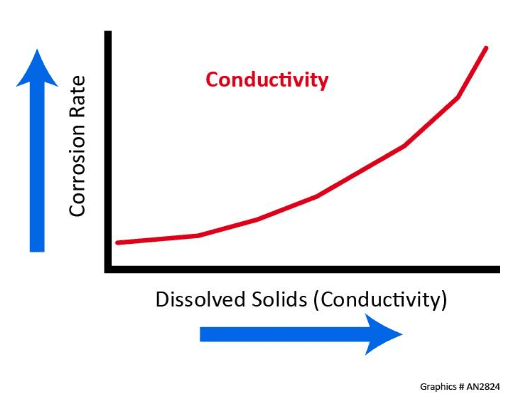
Envisager l’utilisation d’eau de refroidissement dans un système de recirculation ouvert dont la conductivité est de 2 750 μS/cm et comparer cette valeur à l’eau pure avec une conductivité théorique de 0,055 μS/cm. L’eau de refroidissement est 50 000 fois plus conductrice que l’eau pure.
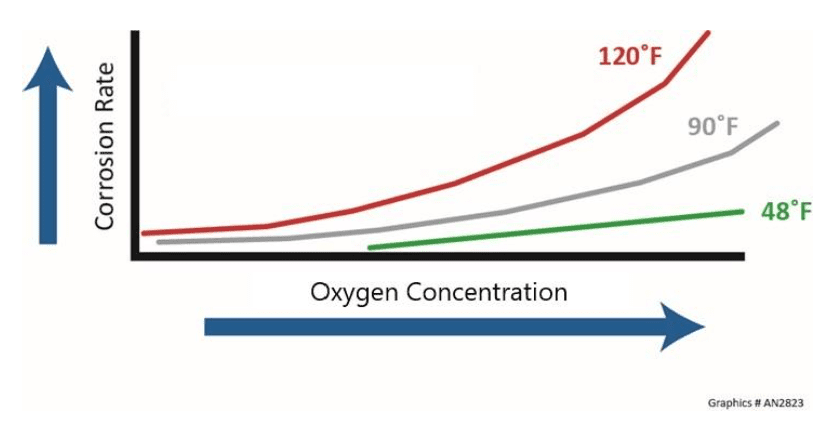
Température et concentration en oxygène dissous
La température a généralement un impact significatif sur le taux de corrosion, et une règle générale suggère que le taux double avec chaque augmentation de 18 °F (10 °C) de la température du liquide. De plus, le taux de corrosion doublera, en général, avec chaque augmentation de 10 à 20 °F de la température de la peau métallique. Certains procédés peuvent produire tellement de chaleur que le gaz d’oxygène moléculaire peut se former sous les dépôts, influençant de manière exponentielle le processus de corrosion. L’effet de la température et de la concentration d’oxygène sur les taux de corrosion est illustré ci-dessous.
Envisager l’utilisation d’eau de refroidissement dans un système de recirculation ouvert dont la conductivité est de 2 750 μS/cm et comparer cette valeur à l’eau pure avec une conductivité théorique de 0,055 μS/cm. L’eau de refroidissement est 50 000 fois plus conductrice que l’eau pure.
Vitesse de l’eau
La vitesse du fluide élevée et faible peut être problématique en ce qui concerne la corrosion. Des vitesses élevées peuvent balayer les produits de corrosion (qui protègent parfois le métal sous-jacent), favorisant ainsi une corrosion supplémentaire. Des vitesses excessives peuvent également provoquer l’érosion et la corrosion, comme décrit plus loin dans cette section. Par contre, un faible débit ou des conditions stagnantes peuvent potentiellement améliorer la corrosion, car cela peut maintenir les ions corrosifs en contact à long terme avec le métal. Une ligne directrice commune pour le débit linéaire à travers la tuyauterie et les tubes d’échangeur de chaleur est de 5 à 10 pieds par seconde (fps), ce qui fournit un équilibre entre le contrôle de la corrosion/dépôt et les coûts des matériaux. Cependant, chaque projet nécessite une analyse minutieuse pour optimiser la conception et les débits de la tuyauterie et de l’équipement. Par exemple, si, pour une raison quelconque, un métal mou est nécessaire pour une application, un débit plus faible peut être nécessaire pour minimiser l’érosion.

LA RAFFINERIE AMÉLIORE LE CONTRÔLE DE LA CORROSION ET RÉDUIT LES COÛTS D'EXPLOITATION AVEC LE FLEXCORR .
Types de corrosion
Nous allons maintenant examiner les types de corrosion les plus courants dans les systèmes de refroidissement. Certains, comme la corrosion générale, permettent souvent une longue durée de vie du matériau et peuvent être contrôlés par des traitements simples. D’autres, comme les piqûres, sont connues pour causer des pénétrations dans les parois traversantes des tuyaux et d’autres équipements en quelques mois, voire quelques semaines. Des arrêts coûteux de l’unité et le remplacement du matériel peuvent en résulter.
Corrosion générale
Avec la corrosion générale, les anodes et les cathodes sur le décalage métallique se développent constamment et les sites de corrosion localisés ne se développent pas. La durée de vie du métal peut être assez longue lorsque la corrosion générale est le seul problème. Les figures ci-dessous illustrent deux exemples de corrosion générale.

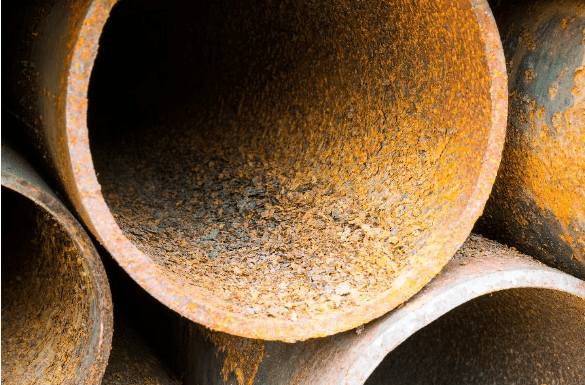
La sélection des matériaux et les dimensions telles que l’épaisseur de la paroi du tuyau sont souvent basées sur une espérance de vie de 30 ans selon les calculs généraux de corrosion. Par exemple, considérez un tuyau de 6 po de diamètre, Schedule 40 avec une épaisseur de paroi de 0,28 po. Lorsqu’environ 50 % de la paroi du tuyau est corrodée, le tuyau peut être presque défectueux et doit être réparé ou remplacé. L’unité commune pour le taux de corrosion est de mils par an (MPY), où un mil est d’un millième de pouce. Dans cet exemple, un taux de corrosion général, et non déraisonnable, de 4,67 MPY entraînerait une perte de paroi de 50 % après 30 ans. De bons programmes de contrôle de la corrosion minimiseront la corrosion générale, mais comme le décrivent les sous-sections suivantes, la corrosion localisée peut être très destructrice et peut être influencée par une variété de facteurs.
Corrosion par piqûres
Si la corrosion se localise, des anodes permanentes se développeront dans un grand environnement cathodique. Les piqûres, ou des mécanismes similaires que nous observerons, sont le résultat, comme illustré ci-dessous.

Le taux de corrosion peut être le même que celui de la corrosion générale, mais les dommages sont beaucoup plus graves en raison de la pénétration rapide du métal aux anodes. De nombreux mécanismes ou conditions peuvent déclencher le piquage. Les dépôts de solides sur l’acier peuvent produire des zones appauvries en oxygène. Ces taches sont anodiques pour nettoyer l’acier. Les colonies microbiologiques peuvent faire de même et peuvent également libérer des composés corrosifs par le biais de processus métaboliques. Nous explorerons ce phénomène un peu plus tard. De mauvaises techniques de soudage peuvent modifier la composition chimique du métal à l’emplacement de soudure et augmenter la sensibilité à la corrosion.
Un phénomène commun avec l’acier au carbone est l’accumulation de produit de corrosion (rouille) sur la fosse.
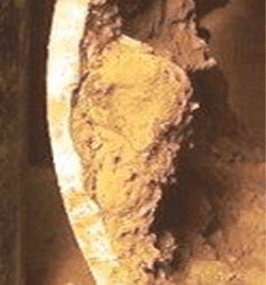
Le liquide emprisonné en dessous peut subir des réactions qui augmentent l’acidité, ce qui augmente le potentiel de corrosion.
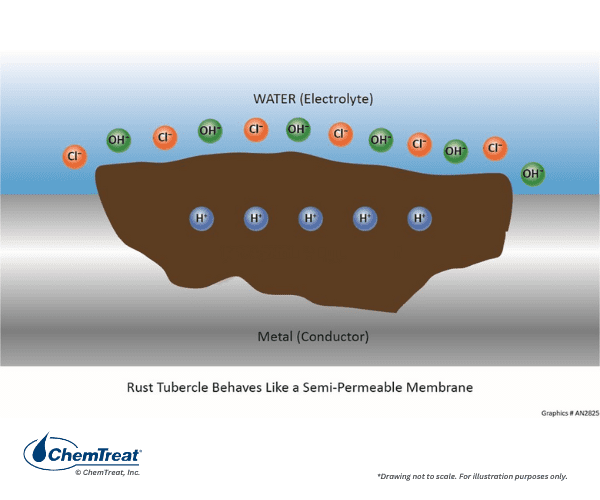
Les chlorures ou autres anions se diffusent dans la fosse pour tenter de maintenir la neutralité de la charge, mais les conditions acides demeurent souvent. Les dépôts au-dessus de la fosse empêchent les inhibiteurs de corrosion de l’eau en vrac de repassiver la surface métallique à l’intérieur de la fosse.
Les figures suivantes illustrent des exemples de piqûres supplémentaires.



7.11a est un piqûre qui a pénétré le bassin métallique d’une tour de refroidissement. 7.11b montre les produits de corrosion à l’extérieur d’un tuyau qui a été généré par une attaque interne et qui a entraîné une pénétration dans le mur traversant. 7.11c illustre une autre pénétration dans le mur traversant.
Pour les fosses en acier au carbone couvertes ou remplies de produits de corrosion, si le matériau est noir (généralement en raison de la présence de magnétite (Fe3O4), la fosse est active et le processus de corrosion est en cours.
Le piquage au chlorure des aciers inoxydables est un problème fréquent, y compris dans les systèmes de recirculation ouverts où l’évaporation de la tour de refroidissement (voir le chapitre 6) augmente la concentration en solides dissous. Les deux aciers inoxydables (SS) les plus populaires pour les tubes de condenseur de surface de vapeur sont 304 et 316, et souvent l'un ou l'autre est spécifié pour un projet sans que les concepteurs ne donnent beaucoup, le cas échéant, pensé aux concentrations de chlorure que les matériaux verront. La difficulté est que les aciers inoxydables forment un revêtement d’oxyde qui protège le métal de base, mais le chlorure en concentration suffisante pénétrera la couche d’oxyde et initiera la piqûre. Pendant des années, les niveaux maximums de chlorure recommandés pour ces aciers variaient de 500 ppm pour 304 SS à 3 000 ppm pour 316L SS à température ambiante. Des recherches ont par la suite montré que ces limites étaient trop élevées, et un expert en matériaux noté suggère 100 et 400 ppm, respectivement, pour les tubes propres. Notez l’accent mis sur des tubes propres. Les dépôts exacerbent le potentiel de corrosion. De plus, les températures au-dessus de la température ambiante influencent grandement le potentiel de fissuration par corrosion sous contrainte (SCC) induite par le chlorure des aciers inoxydables austénitiques, ce qui peut être très préoccupant dans les industries telles que le raffinage du pétrole qui ont de nombreux échangeurs de chaleur pour la production de nombreux composés. Le CS est examiné sous peu.
La résistance aux piqûres de chlorure est souvent un facteur principal dans la sélection des matériaux d’échangeur de chaleur. Un guide bien connu est le tableau des nombres équivalents de résistance au piquage (PREN), comme illustré ci-dessous.
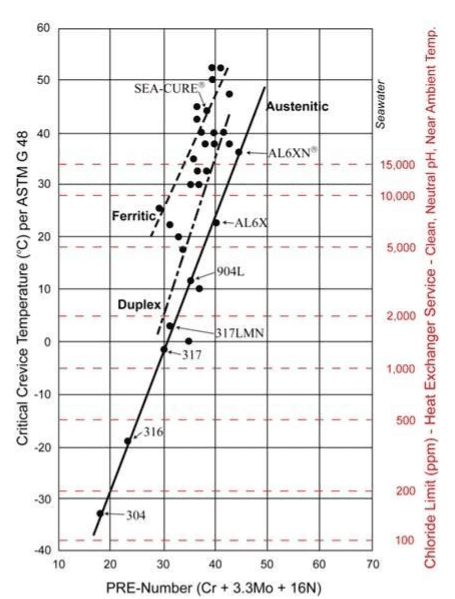
Les matériaux à alliage plus élevé que la série austénitique 300 en acier inoxydable sont recommandés pour les eaux de refroidissement avec des chlorures appréciables. Pour les eaux très saumâtres et l’eau de mer, les alliages ferritiques et superferritiques, par exemple SEA-CURE®, sont souvent nécessaires.
Corrosion de la croûte
La corrosion par gravité est un mécanisme qui se développe aux crevasses des joints mécaniques, par exemple, les joints de bride, les extrémités roulées des tubes, les connexions de boulons ou à d’autres endroits, y compris les limites des dépôts. La figure 7.13 montre un exemple de corrosion des crevasses.

L’eau stagne à ces endroits et peut devenir épuisée en oxygène, ce qui rend les crevasses anodiques à l’autre métal. Tout comme les piqûres, la corrosion est localisée.
Souvent, la corrosion des crevasses n’est découverte qu’après une panne prévue ou si la corrosion cause une défaillance de l’équipement. La meilleure approche pour éviter la corrosion des crevasses est d’éliminer les crevasses. Un traitement chimique approprié minimisera le dépôt et la formation de crevasses à partir de cette source, mais pour l’équipement couplé mécaniquement, des précautions sont nécessaires dans la phase de conception pour éliminer les crevasses lorsque cela est possible. Par exemple, l’installation de joints étanches à l’eau aux raccords à bride peut être une solution dans certaines applications.
Piqûres influencées par le manganèse
Un phénomène qui a touché de nombreux échangeurs de chaleur refroidis à l’eau est la piqûre influencée par le manganèse, avec de nombreux problèmes signalés le long de la rivière Ohio. Même à des concentrations aussi faibles que 0,02 ppm, le manganèse dissous dans l’eau de refroidissement peut être oxydé en dioxyde de manganèse (MnO2) par chloration. Un mince revêtement semblable à un vernis apparaîtra sur les surfaces de l’échangeur de chaleur.

Les dépôts de manganèse sont fortement cathodiques pour le métal sous-jacent et peuvent causer une corrosion localisée grave.

Les aciers inoxydables 304 et 316 sont très sensibles à la corrosion des dépôts de manganèse, mais ils peuvent également affecter le laiton d’admiration, le laiton d’aluminium et le cupro-nickel. L’attaque est probable si la teneur en manganèse du gisement dépasse cinq pour cent. Une concentration supérieure à 20 pour cent entraînera des piqûres graves.
Les autres facteurs qui induisent la formation de MnO2 comprennent un pH élevé, l’aération et parfois des influences catalytiques par la surface métallique elle-même. De plus, la couche de MnO2 peut être oxydée au permanganate (MnO4) par le chlore. Le permanganate dissout le métal de base et, dans le processus, est réduit à MnO2. Le cycle se répète lors de chaque chloration. La corrosion par le manganèse n’est apparemment pas un problème majeur avec l’acier doux, possiblement parce que d’autres produits de corrosion empêchent le manganèse de former un dépôt uniforme et dense.
Les méthodes de dépôt et de contrôle de la corrosion du manganèse comprennent la limitation ou l’élimination de l’alimentation en biocide oxydant (potentiellement en passant à des biocides non oxydants) et l’application d’un programme efficace de stabilisation du manganèse. La sélection de matériaux résistants à la corrosion dans la phase de conception est une autre approche.
Corrosion par érosion
Si l’eau de refroidissement contient des solides en suspension importants, des bulles de gaz ou si la vitesse est tout simplement trop élevée, le liquide circulant peut décaper la couche d’oxyde protectrice sur les métaux et permettre une corrosion continue.

Par exemple, dans une étude de débit de l’eau de mer sur l’acier doux, les taux de corrosion suivants ont été mesurés :
Tableau 7-A : Vitesse linéaire et taux de corrosion de l’eau de mer sur l’acier doux
| Vitesse linéaire (pi/s) | Taux de corrosion (mi/gal) |
| 1 | 7 |
| 4 | 15 |
| 17 | 35 |
Les métaux mous comme le laiton Admiralty sont les plus sensibles à l’érosion, en particulier aux perturbations du débit comme l’extrémité d’entrée des feuilles tubulaires de l’échangeur de chaleur. Ces problèmes doivent être pris en compte pendant la conception du projet.
Cavitation
La cavitation est un type spécifique de corrosion par érosion qui affecte le plus souvent les turbines de la pompe centrifuge.

Si une pression insuffisante est disponible à l’aspiration de la pompe (la pression d’entrée est appelée « tête d’aspiration positive nette (NPSH) »), des bulles dans l’eau peuvent s’effondrer. Les bulles qui s’effondrent peuvent générer de très grandes forces localisées qui décapent l’oxyde protecteur et endommagent le métal. D’autres emplacements potentiels de cavitation comprennent la décharge de la vanne, les régulateurs, les orifices ou d’autres emplacements de chute de pression. Les problèmes potentiels de cavitation doivent être abordés lors de la phase de conception du projet pour s’assurer que les pompes et autres équipements ont une pression de refoulement suffisante.
Fatigue, fatigue liée à la corrosion et craquelures liées à la corrosion par le stress
Presque tous les métaux utilisés dans des applications conventionnelles telles que la tuyauterie, les échangeurs de chaleur et d’autres équipements ne sont pas un seul cristal, mais, selon la référence 1, « sont composés d’une collection de nombreux petits cristaux ou grains ». La structure du grain a un impact énorme sur la métallurgie et plusieurs mécanismes de corrosion.

Un exemple simple de fatigue est facilement démontré en pliant un trombone ou un morceau de fil d’avant en arrière à plusieurs reprises jusqu’à ce qu’il se fracture. Les fractures s’alignent souvent le long des limites du grain. L’équipement de l’usine qui effectue des cycles de charge répétés peut souffrir de fatigue. La corrosion commence généralement par des microfissures qui grossissent avec le temps. Les recherches suggèrent que la fatigue peut également se produire fréquemment dans tous les grains, c.-à-d. transgranulaires.
La fatigue peut être accélérée si le métal est immergé dans un environnement corrosif, même dans les eaux de traitement de base. Au fur et à mesure que les fissures se développent, les produits de corrosion (souvent seulement les oxydes du métal de base) peuvent s’accumuler dans les fissures et exacerber la croissance des fissures. Il s’agit de la fatigue causée par la corrosion.
D’autres mécanismes de corrosion intergranulaire au-delà de la fatigue peuvent être assez problématiques, car une perte de métal relativement faible peut causer une réduction disproportionnée de la résistance du métal.

La corrosion intergranulaire est une attaque localisée qui se produit aux limites du grain, où relativement peu de perte de métal peut causer une réduction disproportionnée de la résistance du métal. Le mécanisme le plus connu est le fissurage par corrosion sous contrainte (SCC), et il peut être particulièrement problématique dans les applications à haute température. (Voir le chapitre 4).
Le SCC nécessite un certain type de contrainte métallique, mais où la contrainte peut être interne ou appliquée.
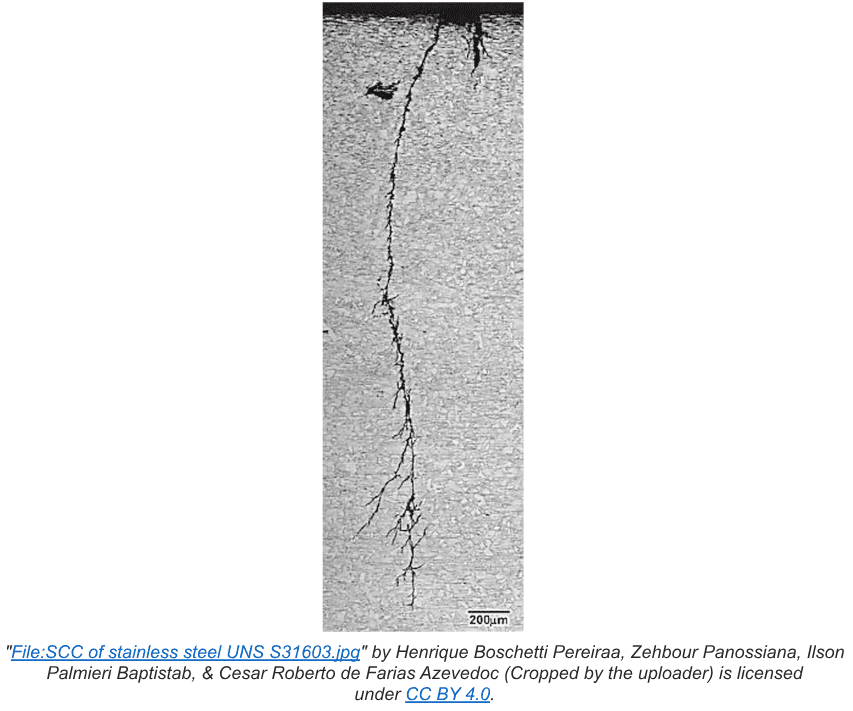
Le stress interne apparaît généralement pendant le processus de fabrication. Un exemple courant de contrainte interne appliquée est le travail à froid ou le roulement à froid de l’acier pour le façonner pour une spécification particulière. Le tuyau cousu est un exemple bien connu. Le traitement thermique et la recuit sont souvent utilisés pour soulager le stress causé par le travail à froid.
Les points faibles générés par le stress peuvent se transformer en microfissures qui deviennent ensuite sensibles aux agents corrosifs dans l’eau. L’exemple le plus connu est peut-être le CS d’acier inoxydable induit par le chlorure. Aux points de stress, des anodes se développent, entourées de métal non stressé qui sert de cathode.
Les emplacements courants pour le SCC dans les systèmes d’eau de refroidissement comprennent les filets coupés en écrous et boulons, les tubes roulés au niveau des feuilles tubulaires, les trous percés ou perforés dans la tuyauterie du distributeur et les coudes de tuyau où le métal a été travaillé mécaniquement.
Le CS peut être atténué par le soulagement du stress dû au traitement thermique. Cependant, il n’est pas toujours pratique de soulager la chaleur à chaque emplacement potentiel. Lors des inspections de l’équipement, les matériaux soumis à des contraintes identifiées ou soupçonnées doivent être particulièrement attentifs pour déterminer l’efficacité des programmes de traitement chimique dans la réduction de la corrosion.
La sensibilité à la corrosion intergranulaire et de contrainte est souvent plus élevée aux soudures. Pour l’acier allié au chrome, le processus de soudage peut causer la précipitation des carbures de chrome qui génèrent des taches de chrome épuisées dans le métal. Ils deviennent anodiques pour le métal de base et deviennent sensibles à la corrosion localisée. De nombreuses défaillances se sont produites aux joints de soudure dans les systèmes d’eau de l’usine. Il est également important de choisir le bon matériau de remplissage de soudure. Un problème sous-reconnu est l’utilisation d’un matériau de remplissage ayant des propriétés de dilatation thermique différentes de celles du métal de base. Une fracture mécanique peut en résulter.
Corrosion galvanique
La corrosion galvanique se produit lorsque deux métaux différents sont en contact physique dans l’environnement d’eau de refroidissement. Revenez au tableau 7-1, qui décrit les potentiels électrochimiques de plusieurs des métaux les plus courants pour l’infrastructure de l’eau de refroidissement. Si deux métaux sont couplés, plus les deux deviennent réactifs, moins ils deviennent réactifs. Plus la séparation du potentiel électrochimique est importante, plus le taux de corrosion potentiel est élevé. Un exemple classique est illustré dans la figure ci-dessous.

Parfois, des cellules galvaniques peuvent se développer selon les réactions chimiques qui se produisent dans le liquide. Le mécanisme de corrosion par piqûres de manganèse décrit précédemment en était un exemple. Un autre mécanisme commun se produit dans les systèmes qui ont à la fois des alliages d’acier et de cuivre, même s’ils ne sont pas physiquement connectés. Si les conditions permettent une certaine corrosion du cuivre, le cuivre peut s’appliquer sur l’acier et produire une cellule de corrosion galvanique.
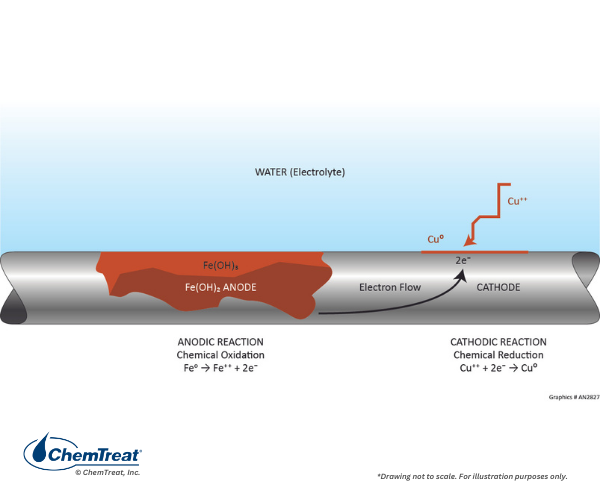
Cet exemple fournit une excellente application des données du tableau 7.1 en examinant la réaction par le biais des potentiels de demi-cellules.
Fe → Fe2+ + 2e–
Cu2+ + 2e– → Cu
V0 = 0,440 V
V0 = 0,340 V
E0 = 0,780 V
Un fort potentiel de conduite existe pour cette réaction.
La solution idéale à la corrosion galvanique est de ne pas avoir de métaux mixtes dans les systèmes de refroidissement, mais cet arrangement est généralement peu pratique. Une clé est d’avoir un faible rapport entre le matériau le plus électronégatif, p. ex., le cuivre, dans un système avec une quantité beaucoup plus importante du matériau électropositif, p. ex., l’acier. Bien que la corrosion galvanique puisse toujours se produire, le rapport anode/cathode élevé garantit que l’attaque est répartie sur une grande zone et ne cause pas de dommages graves.
Dans d’autres cas, des raccords spéciaux peuvent être utilisés pour séparer physiquement différents métaux.

Déallouer
Une forme unique de corrosion qui se produit parfois dans les tubes de l’échangeur de chaleur est déallouante. L’exemple le plus courant est la dézincification du laiton d’admiration. Le laiton Admiralty contient 70 % de cuivre, 29 % de zinc et une petite quantité d’étain, ce dernier pour aider à minimiser la dézincification. Malgré cela, la dézincification peut toujours se produire. La figure 7.24 montre un tube d’admiration qui a subi une perte de zinc.

Deux types de dézincification ont été postulés : la prise et la couche. Dans les deux cas, le zinc se détache du substrat métallique, ce qui entraîne un cuivre poreux et cassant qui n’est pas structurellement solide. Visuellement, l’attaque apparaît comme un timbre rouge terne sur le cuivre jaunâtre.
Le mécanisme réel de dézincification est toujours très débattu. Une théorie est que les deux métaux se corrodent, avec redépôt de cuivre. L’autre est que le zinc sort sélectivement de l’alliage. Les conditions suivantes peuvent améliorer la dézincification :
- Chlorures > 350 ppm
- Résidus halogènes > 1 ppm
- TDS > 3 500 ppm
- pH > 8,3
- Température > 120 oF
Un autre mécanisme de délocalisation qui a été signalé en de rares occasions est la dénickélisation des alliages cuivre-nickel. Ce phénomène semble être assez peu fréquent et ne sera pas abordé plus en détail dans ce livre.
Corrosion induite (influence) microbiologiquement
La corrosion induite microbiologiquement (CMI) est un processus dans lequel les micro-organismes déclenchent, facilitent et/ou accélèrent les réactions de corrosion. Les systèmes de refroidissement offrent un environnement idéal pour les micro-organismes afin d’établir des colonies et de former des dépôts visqueux semblables à de la boue. (Des détails supplémentaires sont fournis dans la section Contrôle microbiologique plus loin dans ce chapitre.) En premier lieu, les dépôts peuvent causer la corrosion différentielle de l’oxygène, comme indiqué précédemment. Au-delà de ce problème, cependant, les processus métaboliques de certains microbes génèrent des composés qui attaquent directement les métaux. Par exemple, les bactéries oxydant le fer comme la gallionelle produisent du chlorure ferrique, connu pour accélérer la piqûre. Les bactéries qui réduisent le sulfate (SRB) comme les désulfovibrio désulfuricans extraient l’oxygène du sulfate (SO4) pour produire du sulfure d’hydrogène (H2S). Les sulfures sous presque toutes les formes sont assez corrosifs pour de nombreux métaux.
La CMI peut laisser des entailles lisses dans la surface métallique comme illustré à la figure 7.25 ci-dessous, et elle peut également causer des piqûres rapides de certains matériaux, y compris les aciers inoxydables.

Corrosion des métaux non ferreux et non métalliques
Jusqu’à présent, une grande partie de la discussion portait sur la corrosion de l’acier, car les aciers constituent la majeure partie du métal dans les systèmes de refroidissement. Cependant, d’autres matériaux métalliques et non métalliques sont souvent présents, et ils peuvent également souffrir de corrosion. Les sections suivantes examinent les problèmes de corrosion pour les matériaux les plus courants.
Alliages de cuivre
Les alliages de cuivre sont souvent le deuxième matériau le plus courant dans les systèmes d’eau de refroidissement, généralement pour les tubes d’échangeur de chaleur. Les alliages de cuivre ont une conductivité thermique plus élevée que l’acier, sont naturellement toxiques pour de nombreuses espèces aquatiques et sont plus résistants que l’acier à certains mécanismes de corrosion.
Comme l’indique le tableau 7.2, le cuivre est noble par rapport à l’ion hydrogène et n’a donc pas tendance à se corroder dans les acides. Cependant, l’oxygène est plus réactif que le H+, et dans les systèmes d’eau de refroidissement, les alliages de cuivre développeront initialement une multicouche d’oxyde cuivreux (Cu2O) de porosité variable, où le cuivre existe dans l’état de valence +1.
- 2Cu + O2 → Cu2O | Éq. 7-9
Au fil du temps, et en présence continue d’oxygène ou d’autres agents oxydants, une oxydation supplémentaire de la couche d’oxyde cuivreux externe peut avoir lieu pour produire une couche d’oxyde cuivreux (CuO) grisâtre-noir.
- Cu2O + ½O2 → 2CuO | Éq. 7-10
Dans certains cas, la couche d’oxyde de cuivre peut rester protectrice, mais certains composés, et plus particulièrement l’ammoniac, peuvent être très corrosifs. Il dissout les ions cupriques à travers un mécanisme connu sous le nom de liaison orbitale.
- Cu2+ + 4NH3 → Cu(NH3)42+ (aq) | Éq. 7-11
Pour les eaux d’appoint contenant une concentration importante d’ammoniac, p. ex., l’effluent de l’usine de traitement des eaux usées, la corrosion du condenseur en alliage de cuivre ou d’autres tubes d’échangeur de chaleur peut être une préoccupation importante. La corrosion des alliages de cuivre a parfois été problématique dans les systèmes de vapeur où l’ammoniac est utilisé pour le contrôle du pH des condensats et de l’eau d’alimentation.
Une autre impureté qui peut causer d’énormes dommages aux alliages de cuivre (et à d’autres métaux) est le sulfure, qui, comme on l’a noté, peut provenir de colonies microbiologiques contenant des bactéries sulfatantes.
- Cu2+ + H2S → CuS↓ + H2↑ | Éq. 7-12
D’autres sources potentielles de sulfure comprennent l’eau qui a été autorisée à devenir septique et les fuites de processus dans les raffineries et les usines similaires. Un exemple surprenant s'est produit il y a plusieurs années, lorsque les 90 à 10 tubes Cu-Ni vieillissants dans un condenseur de surface de vapeur ont été remplacés, pour que les nouveaux tubes tombent en panne suite à une attaque par piqûre dans les 18 mois suivant la mise en service. L’enquête a révélé que le fabricant du tube utilisait un lubrifiant contenant du sulfure, mais n’a pas retiré le composé avant son expédition à l’usine. Les dépôts de sulfure n’ont pas été éliminés, mais plutôt enfouis dans le métal dans des milliers de taches.
Un produit de corrosion du cuivre différent, mais très reconnaissable, est le carbonate de cuivre (CuCO3). Ce verdigris bleuâtre/vert (aussi appelé patine) est souvent vu sur les structures en alliage de cuivre vieilli comme la toiture, les plaques et les statues, et fait généralement partie de la conception architecturale des structures.

La chimie de base est :
- 2Cu + H2O + CO2 + O2 → Cu(OH)2 + CuCO3↓ | Éq. 7-13
La couleur peut varier quelque peu selon le degré d’hydratation du film.
Zinc
En se référant à nouveau au tableau 7.2, le zinc est anodique pour presque tous les métaux, sauf le magnésium et l’aluminium. Contrairement à ces deux métaux, cependant, il ne forme pas une couche d’oxyde super forte. Au lieu de cela, grâce au processus de galvanisation où un revêtement de zinc est appliqué sur les surfaces en acier, le zinc sert d’anode sacrificielle au fer.
La galvanisation est utilisée depuis 1742. La galvanisation moderne se fait par trempage à chaud, bien que les procédés continus tels que la galvanisation des usines lourdes soient courants pour l’acier en tôle. L’immersion à chaud est un processus par lots dans lequel les composants en acier nouvellement fabriqués sont immergés dans une solution de zinc fondu pendant une durée prescrite. Le zinc fusionne avec la surface en acier. L’épaisseur du revêtement est directement proportionnelle à la durée d’immersion, qui peut être ajustée par taille de composant et application.
Les matériaux récemment galvanisés sont souvent brillants et très réfléchissants, comme le montre la figure 7.27.

Cependant, tous les revêtements galvanisés ne sont pas brillants. Certains éléments de l’acier, comme le silicium et le phosphore, peuvent accélérer la croissance des couches d’alliage de zinc-fer. Cela peut produire un revêtement galvanisé fini entièrement composé d’alliage de zinc et de fer.

La formation de la patine protectrice gris foncé (fini de surface) commence par le développement d’une fine couche d’oxyde de zinc à la surface. Dans des conditions appropriées, ces oxydes incorporeront une couche de carbonate de zinc de base au contact de l’eau. Après l’exposition initiale à l’eau et au dioxyde de carbone (qui peut être obtenue en permettant aux nouveaux composants du système de refroidissement galvanisé de connaître des conditions extérieures sur une période de plusieurs mois), les composants galvanisés industriels peuvent ensuite être exposés à l’eau du système. L’étape de revitalisation nécessite un contrôle minutieux pour produire un composé de carbonate de zinc hydraté dont on pense qu’il contient la formule suivante :
- 3Zn(OH)2∙ZnCO3∙H2O
Cette couche se développe et devient plus protectrice au fil du temps, mais si la chimie de l’eau appropriée n’est pas établie pendant le processus de conditionnement, un matériau non protecteur appelé « rouille blanche » peut se former. De plus amples détails concernant la chimie de passivation du zinc sont examinés dans une section ultérieure sur le démarrage préopérationnel des systèmes de refroidissement.
Les composants galvanisés sont courants dans de nombreux emplacements de tours de refroidissement, même si la structure de support principale est en bois ou en plastique.
De nombreuses tours de refroidissement commerciales de taille modeste peuvent être entièrement fabriquées en acier galvanisé.

Aluminium
En repensant au tableau 7.1 au début de ce chapitre, il est difficile d’imaginer que l’aluminium et, dans certains cas, le magnésium, conviennent à des fins de construction, car ils sont si réactifs. La clé est que la réactivité élevée induit la formation d’une couche d’oxyde serrée qui protège le métal en dessous. Les produits typiques en aluminium comprennent les moules à injection, les blocs-moteurs, les radiateurs, les échelles et les passerelles de tours de refroidissement, les mains courantes, les pales de ventilateur et d’autres composants similaires. L’aluminium résiste à la corrosion atmosphérique et cet aspect, associé à son poids léger, en fait un excellent matériau pour les composants d’avion.
L’aluminium est un matériau amphotérique, ce qui signifie qu’il se corrodera à un pH faible et élevé.
L’aluminium peut se corroder dans les eaux alcalines avec du phosphate. De plus, étant donné que l’aluminium est plus actif électrochimiquement que l’acier, il se corrode lorsqu’il est couplé à l’acier dans les environnements d’eau de refroidissement.
Problèmes de corrosion non métalliques
Cette section examine brièvement deux problèmes de corrosion non métalliques principalement liés aux grandes tours de refroidissement. Il s’agit de la dégradation du bois (qui peut servir de structure de support de tour) et du béton, qui est un matériau commun pour les bassins de tour de refroidissement.
Dégradation du bois
Pour les nombreuses grandes tours de refroidissement qui ont encore des composants en bois, cette section fournit un aperçu des influences de dégradation. La détérioration du bois peut généralement être classée en trois catégories : physique, chimique et microbiologique.
- Physique
- Températures d’eau de refroidissement excessivement élevées supérieures à 140 °F
- Dommages causés par la glace
- Érosion
- « Pourriture en fer » des fixations métalliques adjacentes rouillées (Figure 7.30)
- Dommages cycliques humides/secs
- Stress mécanique (Figure 7.31)


- Produit chimique
- Concentrations élevées de chlore dépassant 1,0 ppm, ce qui peut déligner le bois. La lignine est le liant des fibres de cellulose dans le bois, mais elle peut être détruite par une teneur élevée en halogène.
- Lignes d’injection de produits chimiques placées près de structures en bois. Si ces lignes présentent des fuites, les produits chimiques concentrés peuvent attaquer le bois.
- Des valeurs de pH élevées supérieures à 9,0. Un pH élevé peut être particulièrement destructeur lorsqu’il est combiné à des concentrations élevées de chlore. Les programmes de traitement de l’eau de refroidissement sont généralement conçus pour fonctionner sous ce pH.
- Biologique (principalement lié aux champignons)
- Les champignons sont responsables de la carie et de la destruction du bois
- Les champignons existent sous forme de levures (unicellulaires) et de moisissures (multicellulaires-filamenteuses)
- Les champignons se développent dans une plage de pH légèrement acide de 5,5 à 6,5.
- Les champignons ont besoin de niveaux élevés de carbone organique, comparativement aux bactéries. Il s’ensuit que la prolifération des champignons d’eau en vrac est relativement rare, à moins qu’une contamination organique ne soit présente, peut-être à cause de fuites de processus d’hydrocarbures ou si l’approvisionnement en maquillage est un effluent municipal de l’usine de traitement des eaux usées.
La discussion suivante décrit les trois principaux types de pourriture fongique du bois.
Rot brun : La pourriture brune, également connue sous le nom de pourriture sèche, est plus courante dans les bois tendres et attaque sous la couche d’agent de conservation appliquée pendant la fabrication. Les champignons recherchent la cellulose, laissant derrière eux des lignines de couleur foncée. Il peut pénétrer profondément dans le bois.

Roteau blanc : La pourriture blanche est plus courante dans les bois durs et attaque les lignines. Elle progresse plus lentement que la pourriture brune. La surface du bois devient douce et fictive et semble blanchie. La pourriture blanche peut être contrôlée par des traitements fongicides de surface aux premiers stades de croissance. Le fongicide pénètre lentement dans le bois.

Roteau souple : La pourriture molle ne se produit que dans les zones lavées à l’eau et est confinée à la surface aux premiers stades. L’attaque est plus lente que la pourriture blanche ou brune. La surface semble fissurée et de couleur claire lorsqu’elle est sèche. La pourriture molle peut être contrôlée par un traitement microbiologique diligent de l’eau de refroidissement.

Corrosion du béton
Le béton était le matériau de construction des grandes tours de refroidissement hyperboliques des centrales nucléaires et de certaines centrales au charbon au siècle dernier. Pratiquement aucune tour hyperbolique n’a été construite au cours des dernières décennies et ne sera pas considérée plus loin ici. Cependant, bon nombre des grandes tours de tirage mécaniques de l’industrie lourde et de l’électricité ont des bassins en béton renforcé.

Le béton est solide, coulé sur place et peut avoir une longue durée de vie. Au fil des ans, un problème courant est survenu dans les usines où l’acide sulfurique est utilisé pour le contrôle du pH de l’eau de refroidissement et où l’acide n’est pas dilué avant l’injection dans la cuvette. L’acide sulfurique de qualité commerciale (concentration de 93 à 98 %) a une densité presque deux fois supérieure à celle de l’eau et s’enfoncera rapidement dans le plancher de la cuvette s’il n’est pas dilué, où il peut attaquer le béton et les barres de renforcement du béton.
La principale méthode pour minimiser de tels dommages est un système de dilution acide, qui mélange l’acide et l’eau à l’extérieur, avec la distribution du mélange à travers un creux au-dessus du bassin.

Le ciment Portland standard peut être attaqué par les eaux avec une concentration élevée de sulfate (>1 500 ppm), ce qui est possible dans certains systèmes de refroidissement selon l’effet de concentration de la tour. Ce problème peut être résolu lors de la phase de conception avec un calcul minutieux de la qualité de l’eau d’appoint et de la mesure dans laquelle le sulfate se concentrera lorsque la tour sera cyclée à des niveaux normaux. Les conditions peuvent nécessiter l’utilisation de ciment Portland de type V, qui contient une quantité réduite d’aluminate tricalcique, habituellement l’un des principaux composants du ciment standard.
Avant d’examiner les méthodes de contrôle de la corrosion, nous examinerons les principales causes de dépôt et de détartrage dans les systèmes de refroidissement. Les programmes de traitement de l’eau passés et actuels sont généralement des mélanges d’inhibiteurs de corrosion et de dépôts, et par conséquent, la discussion de l’un comprend souvent l’autre.
Mécanismes de dépôt
Outre la formation de tartre, le dépôt de solides dans les systèmes de refroidissement peut se produire par plusieurs mécanismes supplémentaires, notamment :
- Régulation dans les zones à débit restreint
- Contamination par des particules en suspension dans l’air qui pénètrent dans la tour de refroidissement
- Encrassement microbiologique
- Encrassement macrobiologique
- Encrassement causé par la graisse et l’huile ou d’autres contaminants organiques du procédé
- Produits de corrosion provenant d’autres zones du système
Problèmes de débit restreint
Un emplacement à débit restreint commun, décrit au chapitre 6, est le remplissage de la tour de refroidissement.
Le remplissage de feuil à haute efficacité offre un excellent transfert de chaleur, mais au prix d’une trajectoire de débit tortureuse qui réduit la vitesse de l’eau. Le débit régulé vers les échangeurs de chaleur peut également établir des zones à faible débit qui recueillent des solides. Un article souvent négligé avec de grands systèmes de refroidissement sont des pattes mortes qui peuvent accumuler des matériaux, y compris des microbes.
Particules en suspension dans l’air
Les tours de refroidissement sont de superbes épurateurs d’air, et de nombreux solides peuvent être introduits par ce chemin d’écoulement. L’infiltration de poussière pendant les périodes sèches est un problème courant. Un autre exemple classique avec lequel de nombreux opérateurs sont familiers est l’intrusion de graines de bois de coton et de végétation végétale supplémentaire qui obstrue les crépines et autres équipements.
Encrassement microbiologique
L’eau et l’air sont remplis de microbes qui peuvent potentiellement former des colonies problématiques dans les systèmes de refroidissement. L’encrassement peut se produire très rapidement et potentiellement forcer le déclassement de l’unité ou même l’arrêt de l’équipement dans les jours suivant l’apparition microbienne. La boue protectrice sécrétée par certains microbes emprisonne facilement les solides en suspension qui convertissent le matériau en un produit semblable à la boue.

Encrassement macrobiologique
Un certain nombre de créatures aquatiques qui échappent aux tamis d’entrée d’eau de refroidissement peuvent bloquer les extrémités d’entrée des tubes d’échangeur de chaleur. Ces difficultés ont été particulièrement problématiques dans les condenseurs de surface de vapeur de centrale électrique à passage unique. Certaines des créatures les plus courantes comprennent les palourdes asiatiques, les moules zébrées et même les petits poissons comme l’ombre.
Traiter l’encrassement des contaminants
De nombreuses grandes industries ont de nombreux échangeurs de chaleur. Les fuites de l’échangeur de chaleur peuvent introduire des contaminants dans l’eau de refroidissement qui retourne à la tour. Les huiles et les hydrocarbures lourds qui peuvent enduire l’équipement du système de refroidissement sont particulièrement gênants.
Dépôt de produit de corrosion
La corrosion est problématique en soi, mais la corrosion libère des produits qui se logent ensuite dans d’autres endroits.
Formation de tartre
Le chapitre 1 comprenait une discussion sur les produits de solubilité et sur la façon dont les précipitations de matières solides se produisent lorsque divers ions dissous atteignent une limite de solubilité. Il s’agit du mécanisme derrière la formation de tartre dans les systèmes d’eau.

Au chapitre 2, nous avons appris que le précipité le plus courant dans les eaux naturelles est le carbonate de calcium (CaCO3) et comment la chimie des précipitations de CaCO3 peut être utilisée de manière avantageuse dans les réactions d’adoucissement de la chaux pour le traitement de l’eau d’appoint. À l’inverse, la formation indésirable de tartre de carbonate de calcium dans les systèmes d’eau, y compris la plomberie domestique, a affecté l’humanité depuis des années, et dont le traitement a lancé la chimie moderne de contrôle du tartre que nous connaissons aujourd’hui. En bref, presque toutes les eaux naturelles contiennent des ions calcium dissous (Ca2+) et de l’alcalinité bicarbonate (HCO3–). Selon diverses influences, y compris la température, les ions précipiteront de la solution. La réaction suivante est représentative de ce processus.
- Ca2+ + 2HCO3– + chaleur → CaCO3↓ + CO2↑ + H2O | Éq. 7-14
Le carbonate de calcium a trois polymorphes. Le calcite est la forme thermodynamique la plus stable et comprend la plupart des dépôts naturels.

Une forme moins stable est l’aragonite, qui se trouve principalement dans le CaCO3 biosynthétique comme les coquilles et les coraux. La structure finale est la vatérite, qui se produit rarement dans la nature, mais qui joue un rôle transitionnel important dans la formation de carbonate de calcium à partir de la solution.
Le dépôt de carbonate de calcium a été le moteur du développement des premiers programmes pour prédire les tendances de formation de tartre (et de corrosion) des impuretés de l’eau. Ces développements sont soulignés ci-dessous.
Indices de saturation
Indice de saturation de Langelier (LSI)
En 1936, le Dr Wilfred F. Langelier (1886 à 1981) a fait des recherches sur un problème de corrosion dans la tuyauterie d’alimentation en eau de Cleveland, Ohio. Il a appris que la corrosion pouvait être réduite en augmentant le pH de l’eau traitée, mais avec le compromis d’un potentiel accru de tartre de carbonate de calcium.
Il a développé la LSI, un modèle d’équilibre dérivé de l’évaluation théorique de la saturation en carbonate de calcium. On dit qu’une eau est saturée de carbonate de calcium lorsqu’elle ne se dissoudra ni ne précipitera le minéral. Ses calculs pourraient prédire quand le détartrage du carbonate de calcium se produirait en mesurant les concentrations de calcium, d’alcalinité du bicarbonate, de pH et de solides dissous totaux, dans la plage de température commune de l’eau.
L’équation fondamentale est :
- LSI = pH – pHs | Éq. 7 à 15
Où;
- pH = pH réel mesuré
- pH = pH de saturation en carbonate de calcium. Il s’agit d’une valeur calculée où le carbonate de calcium en solution est en équilibre avec le carbonate de calcium précipité.
- pH = (pK2 ‒ pKs) + pCa + pAlk Eq 7-16
- pK2 = log10 négatif de la deuxième constante de dissociation pour l’acide carbonique; pK = log10 négatif du produit de solubilité (Ksp) pour la calcite (CaCO3); pCa = log10 négatif de la concentration en calcium; et pAlk = log10 négatif de la concentration totale d’alcalinité.
- pH = (pK2 ‒ pKs) + pCa + pAlk Eq 7-16
Les valeurs de pK2 et de pK dépendent de la température. À mesure que la technique LSI gagnait en faveur, les chercheurs ont développé des nomographies qui ont permis un calcul rapide de (pK2 - pKs) dans les plages de température typiques à passage unique ou à recirculation ouverte.
La corrélation empirique des calculs est résumée comme suit :
- LSI < 0, le tartre de carbonate de calcium se dissoudra et il existe un potentiel de corrosion de l’acier doux.
- LSI = 0, l’eau est stable en ce qui concerne la formation et la dissolution de carbonate de calcium.
- LSI > 0, le potentiel de formation de tartre de carbonate de calcium augmente avec l’augmentation de la LSI.
Langelier a pu contrôler la corrosion tout en évitant la formation de tartre en ajustant l’alimentation en chaux pour maintenir une plage de +0,5 à +1,0 LSI dans l’eau de la ville.
Pour les systèmes d’eau à passage unique et fermée, l’ajout d’alcalis pour augmenter la LSI à 0,5 ou environ peut minimiser la corrosivité, mais ne pas atteindre des conditions de formation de tartre sévères. Dans un système de refroidissement à recirculation, il peut être possible d’augmenter la LSI ou les autres indices décrits ci-dessous en augmentant les cycles de concentration, ce qui augmente la dureté calcique et l’alcalinité du carbonate.
Indice de stabilité Ryznar (RSI)
En 1944, John W. Ryznar (1912 à 1996) a proposé une modification substantielle à la LSI. Il a découvert qu’il était possible que les eaux à faible dureté et à forte dureté aient la même LSI selon l’alcalinité et le pH connexe. Ryznar a nommé sa relation l’indice de stabilité et a corroboré son IRV avec des données expérimentales. L’équation de Ryznar utilise les mêmes données que la LSI, mais le calcul final est :
- RSI = 2pH – pH | Éq. 7 à 17
La corrélation empirique de l’ISR est résumée comme suit :
- RSI < 6, la tendance au tartre de carbonate de calcium augmente à mesure que l’indice diminue.
- RSI = 6, l’eau est stable en ce qui concerne la formation et la dissolution de carbonate de calcium.
- RSI > 6, le tartre de carbonate de calcium se dissoudra et la corrosion légère de l’acier devient une probabilité croissante avec les valeurs croissantes.
Indice de mise à l’échelle pratique (IPV)
L’indice d’échelle pratique (IPV) a été développé par Paul Puckorius (1930 à 2019), qui, lorsqu’il était jeune, était assistant de Ryznar. Il incorpore un pH calculé de l’eau basé sur la capacité tampon, au lieu d’un pH simplement mesuré. L’équation de l’indice de mise à l’échelle pratique (IPV) est :
- (PSI) = 2(pH) ‒ pHeq | Éq. 7-18
- pHeq = 1,465 x log10 (alcalinité totale) + 4,54 | Éq. 7-19
La corrélation empirique de l’IPV est la même que celle de l’IPV. Au moins un des principaux fabricants de turbines à combustion utilise PSI pour calculer les tendances de tartre pour les refroidisseurs d’air d’entrée. Placer les calculs dans un tableur est simple.
D’autres indices prédictifs sont disponibles, y compris l’indice Oddo-Tomson, l’indice Stiff-Davis, les niveaux de saturation, l’excès momentané et d’autres. Il est utile à ce stade de présenter un autre calcul, l’indice Larson-Skold pour le potentiel de corrosion. Les anions agressifs comme le chlorure et le sulfate sont plus conducteurs électriquement que les anions tampons, le bicarbonate et le carbonate. Dans les années 1950, le Dr T. E. Larson et le Dr R. V. Skold ont étudié la corrosivité des eaux des Grands Lacs et ont développé la formule suivante.
- Indice Larson-Skold = (ppm Cl– + epm SO42- epm HCO3– + epm CO32-) | Éq. 7-20
(epm = équivalents par million)
Ils ont découvert que lorsque le rapport entre les anions forts et les anions faibles était inférieur à 0,2, les anions tampons ont une plus grande influence que les anions corrosifs et peuvent former un film inhibiteur naturel. Cependant, lorsque l’indice s’élève au-dessus de 0,6, la situation est inversée et le potentiel de corrosion est plus élevé. La relation empirique Larson-Skold était spécifiquement basée sur les eaux des Grands Lacs. Bien que des relations similaires puissent être calculées pour d’autres eaux, les prédictions peuvent être différentes.
Bien que certaines entreprises de traitement de l’eau utilisent toujours ces calculs pour une évaluation rapide des tendances de détartrage de l’eau, les méthodes n’ont pas les capacités des programmes informatiques modernes, qui tiennent compte de facteurs supplémentaires, y compris l’effet commun des ions. Des programmes très sophistiqués sont disponibles qui permettent à l’utilisateur d’entrer non seulement les conditions du système et la chimie de l’eau brute, mais aussi les types et les concentrations réels d’inhibiteurs de tartre. Les programmes calculeront la plage normale et les conditions limites pour tout programme de traitement souhaité. Les sections suivantes examinent d’autres mécanismes de mise à l’échelle.
Autres échelles
Selon la chimie de l’eau d’appoint ou la façon dont elle change lorsqu’elle est cyclée dans une tour de refroidissement, d’autres dépôts minéraux sont possibles dans les systèmes de refroidissement. Le tableau 7-2 décrit les plus courants.
Tableau 7-2. Autres dépôts courants de tartre d’eau de refroidissement
| Composé | Formule |
| Gypse | CaSO4∙2H2O |
| Silice | SiO2 |
| Silicate de magnésium | MgSiO3 |
| Phosphate de calcium | Ca3(PO4)2 |
| Fluorite | CaF2 |
Comme nous le verrons, certains problèmes de détartrage, et notamment ceux liés au dépôt de sulfate et de phosphate, sont apparus en grande partie des progrès ou des changements dans les programmes de traitement chimique.
Avant de continuer, un point important doit être mis en évidence concernant les composés minéraux dans le tableau 7-2 par rapport au CaCO3. L’anion contenu dans le carbonate de calcium est le CO3. En termes chimiques alternatifs, le CO3 est la « base conjuguée » de l’acide faible H2CO3. Une discussion sur les acides conjugués et les bases dépasse la portée de ce livre, mais l’idée clé est que les dépôts de carbonate peuvent généralement être éliminés par application d’acide, même sous forme diluée.
- CaCO3 + H2SO4 → Ca2+(aq) + SO42-(aq) + H2CO3 | Éq. 7-21
- H2CO3 �.COLORE CO2↑ + H2O | Éq. 7-22
Par conséquent, si l’acide est fourni en quantités suffisantes avec un contact uniforme, les dépôts de CaCO3 se dissoudront entièrement au fur et à mesure que le carbonate se transforme en dioxyde de carbone. L’apport d’acide sulfurique à l’appoint de la tour de refroidissement était, et dans certains cas, il l’est encore, une méthode courante pour réduire l’alcalinité et réduire le potentiel de formation de tartre CaCO3. Les besoins en acides ne sont souvent pas assez importants pour causer des précipitations de sulfate de calcium, mais le problème ne peut pas être ignoré.
Sulfate de calcium
Un problème parfois problématique est l’entartrage du gypse (CaSO4∙2H2O), influencé par des concentrations élevées de sulfate dans le maquillage ou par le traitement acide pour éliminer le carbonate.
Le sulfate de calcium a une solubilité plus élevée que le CaCO3, comme illustré ci-dessous.
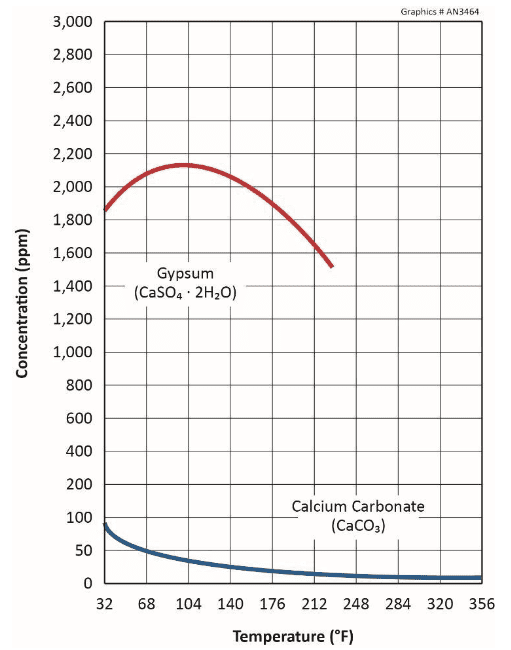
La figure révèle que le gypse présente également une solubilité inverse, mais pas avant que les températures atteignent environ 105 °F.
Une ligne directrice générale courante suggère des limites de 1 200 ppm de calcium (mg/L sous forme de CaCO3) et de 1 200 ppm de sulfate (mg/L sous forme de SO4), ou quelques-uns de ceux-ci, pour empêcher la formation de tartre à des températures normales du système de refroidissement dans l’eau non traitée. Des limites plus élevées peuvent être possibles avec le traitement chimique, mais ces cas doivent être évalués sur une base individuelle.
Phosphate de calcium
Comme nous le verrons plus en détail dans la section suivante, dans les années 1980, un changement majeur dans le traitement chimique des systèmes de recirculation ouverts s’est produit avec l’adoption de la chimie du phosphate inorganique et organique pour le contrôle du tartre et de la corrosion. Soudainement, le dépôt de phosphate tricalcique (Ca3(PO4)2) est devenu un problème majeur dans de nombreuses installations.
Outre le phosphate tricalcique (Ca3(PO4)2), d’autres phases de phosphate de calcium peuvent se former dans l’eau de refroidissement. On suppose souvent que l’hydroxyapatite thermodynamique stable (Ca5(PO4)3(OH)) est un prototype approprié pour la prédiction du tartre. Il semble que pendant la précipitation du phosphate de calcium, le phosphate de calcium amorphe (PCA) se forme d’abord, suivi de la nucléation et de la transformation en phase d’autres composés.
PCA → Précurseur → Phase stable
La solubilité du ou des phosphate(s) de calcium dépend fortement du pH et de la température de la solution. Toutes les espèces présentent une solubilité inverse par rapport à ces deux paramètres. La tendance à l’entartrage du phosphate de calcium dépend également d’autres influences, y compris celles provenant d’autres ions métalliques. Tous les facteurs doivent être pris en compte lors du calcul du potentiel de mise à l’échelle.
Silice/silicates
La chimie aqueuse de la silice est complexe et un certain nombre de précipités peuvent se former selon la température, le pH et d’autres facteurs. Les dépôts potentiels comprennent :
- Silice amorphe dans les sections plus froides du système d’eau
- Silicates métalliques dans des endroits chauds ou chauds ou à un pH élevé
- Minéraux de silicate comme l’argile qui sont en suspension dans l’eau
La silice amorphe est simplement SiO2. De nombreuses eaux de surface contiennent de faibles niveaux (< 15 ppm) de silice, mais certaines eaux souterraines peuvent avoir des concentrations allant de 75 à 80 ppm. À température ambiante, le niveau de saturation en silice est d’environ 150 ppm, de sorte que lorsque la concentration de silice augmente à la saturation et au-dessus, un processus de polymérisation induit la formation de silice colloïdale qui peut se fixer aux surfaces du système. Ce dépôt se produit principalement dans les endroits les plus frais, comme le remplissage de la tour.
Dans la plage de pH d’environ 2,0 à 8,3, la solubilité de la silice est indépendante du pH, mais la silice dissoute se convertit en silicate (SiO3–) lorsque le pH dépasse 8,3. Les silicates précipiteront avec les cations, notamment le magnésium et le calcium. Ces composés présentent une solubilité inverse en ce qui concerne le pH et la température, et s’accumuleront donc d’abord dans des endroits chauds, c.-à-d. des échangeurs de chaleur. Les écailles de silice et de silicate sont très tenaces et difficiles à enlever. Ce sont également des isolateurs puissants qui réduisent considérablement le transfert de chaleur.
Certains programmes de traitement chimique peuvent permettre un fonctionnement avec des concentrations de silice dissoute égales ou peut-être même un peu supérieures à 200 ppm. Cependant, une connaissance approfondie de la chimie de l’eau est nécessaire pour repousser les limites du programme. Par exemple, les ions polyvalents, tels que Zn2+ et Al3+, sont entourés de groupes hydroxyles qui peuvent catalyser la polymérisation de la silice. Parmi tous les cations, le magnésium a le plus grand potentiel d’induire le dépôt de silicate.
La silice dissoute peut être analysée par spectrophotométrie ultraviolette/visible (UV/VIS) par la méthode du molybdate. La mesure totale de la silice, y compris la forme colloïdale où la silice existe sous forme de particules solides, nécessite des techniques plus avancées, telles que la spectroscopie à plasma à couplage inductif (PCI) ou à absorption atomique (AA).
Contrôle des dépôts et de la corrosion
Les méthodes de traitement chimique pour le contrôle de la corrosion et du tartre sont entrelacées depuis des années, et cette section fournit un examen des programmes les plus courants au cours du dernier demi-siècle ou plus, et de la façon dont les méthodes passées et actuelles ont été conçues pour résoudre les deux problèmes.
Revenez à l’équation 7-14. Au milieu du siècle dernier, un programme de traitement extrêmement populaire pour les systèmes de recirculation ouverte était l’alimentation en acide sulfurique pour le contrôle du tartre (pour établir une plage de pH commune de 6,5 à 7,0), avec l’alimentation en chromate disodique (Na2Cr2O7) pour le contrôle de la corrosion. Ce dernier composé fournit des ions chromate (CrO42-) qui réagissent avec l’acier au carbone pour former une couche d’acier pseudo-inoxydable qui peut être très protectrice.
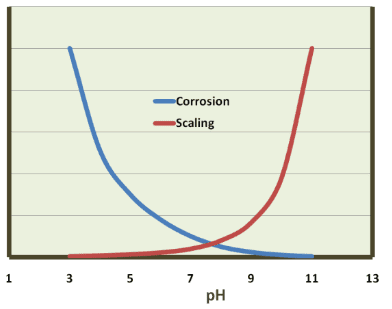
Cependant, dans les années 1970 et 1980, la reconnaissance au réveil de la toxicité du chrome hexavalent (Cr6+) a conduit à une interdiction de décharge de chrome dans l’environnement, ce qui a essentiellement éliminé le traitement au chromate pour les systèmes d’eau de refroidissement ouverts. Le programme de remplacement était très différent, un concept clé étant le fonctionnement à un pH légèrement basique (généralement environ 8,0 ou peut-être un peu plus élevé) pour aider au contrôle de la corrosion. Les produits chimiques de traitement de base sont devenus des phosphates inorganiques et organiques. Mais, comme nous le verrons, cette chimie plus compliquée (par rapport au chromate acide) a augmenté le potentiel de tartre. La figure 7.41 illustre succinctement la relation générale entre la corrosion et le tartre.
La capacité du phosphate à influencer le pH est démontrée par la réaction du phosphate trisodique (Na3PO4, TSP) dans l’eau.
- Na3PO4 + H2O �. TAILLE NaH2PO4 + NaOH | Éq. 7-23
La chimie du TSP est utilisée depuis des décennies pour ajuster le pH dans les générateurs de vapeur haute pression (voir le chapitre 4). Mais les phosphates inorganiques appliqués seuls sur l’eau de refroidissement peuvent induire une formation importante de Ca3(PO4)2, et en effet, lorsque la chimie du phosphate a émergé comme remplacement du chromate acide, le dépôt de phosphate de calcium est devenu très problématique. Par conséquent, des formulations qui comprenaient des polyphosphates, des phosphates organiques (aussi appelés phosphonates), des polymères et souvent une petite concentration de zinc, toutes conçues pour le contrôle intégré du tartre et de la corrosion ont émergé.
La concentration résiduelle optimale en phosphate dépend de facteurs tels que l’indice LSI/RSI/PSI de l’eau, le pH, la température et le type d’autres inhibiteurs dans le programme de traitement. Une trop grande quantité de phosphate peut entraîner un détartrage tricalcique du phosphate sur les surfaces chaudes. Le phosphate peut également précipiter avec du fer et de l’aluminium.
Une plage typique de contrôle de l’orthophosphate est de 6 à 18 ppm.
Polyphosphates
Les polyphosphates contiennent plusieurs atomes de phosphore reliés entre eux par des ponts d’oxygène, comme le montre la figure 7.42. Les polyphosphates contiennent généralement de 3 à 5 unités et ont des atomes d’oxygène chargés négativement qui attirent les cations, y compris le calcium et le fer. Cette attraction séquestre efficacement les cations, les empêchant de former des dépôts.
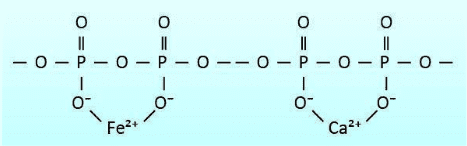
Une « concentration du seuil » est nécessaire pour inhiber l’entartrage du carbonate de calcium lorsque les concentrations sont à des niveaux de saturation. Le polyphosphate se combine également au manganèse. Le tripolyphosphate de sodium ((Na5P3O10), le pyrophosphate de tétrasodium (Na4P2O7) et l’hexamétaphosphate de sodium ((NaPO3)6) ne sont que quelques-uns des polyphosphates. Normalement, 2 à 5 ppm de polyphosphate sont nécessaires dans un programme de traitement. Les polyphosphates seront hydrolysés et retourneront à l’orthophosphate, où divers facteurs tels que le temps de résidence et la température influencent le taux de réversion.
Organophosphates
Les phosphonates inhibent la formation de tartre par adsorption sur les cristaux actifs pour retarder la nucléation et le taux de croissance des cristaux. Les phosphonates agissent également comme des séquestrants qui forment des complexes avec divers cations. Certains phosphonates offrent une protection contre la corrosion, comme décrit brièvement ici et dans la section suivante. Quatre phosphonates courants sont illustrés ci-dessous.
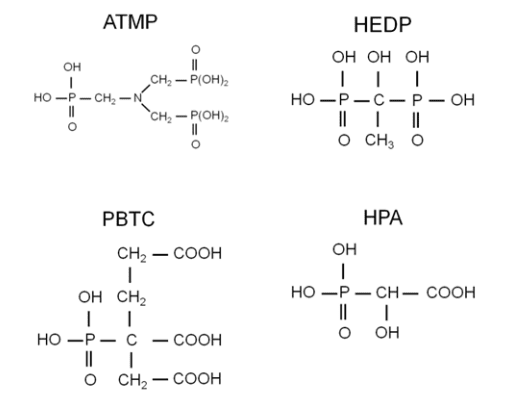
L’ATMP a été le premier phosphonate et a été introduit au début des années 1970 pour le contrôle du tartre de carbonate de calcium. Il a servi de remplacement aux polyphosphates et pourrait étendre les indices de saturation décrits précédemment à :
- LSI de +1,5
- RSI/PSI d’ici ‒1,5
ATMP a démontré des propriétés d’inhibiteur de corrosion équitables à bonnes aux plages de pH alcalines des programmes phosphate-phosphonate (alors neufs). Cependant, l’ATM a une faible tolérance aux biocides oxydants comme le chlore, et il peut également former des précipités de calcium-phosphonate.
Une amélioration est survenue avec la HEDP, qui fonctionne de la même façon que la ATMP, mais qui a une tolérance plus élevée aux oxydants. HEDP a remplacé ATMP dans la plupart des applications. D’autres recherches ont mené au développement du PBTC, qui a une tolérance encore plus élevée pour le chlore et le brome que le HEDP. Notez les groupes d’acide carboxylique (COOH) sur cette molécule, qui reviennent au carboxylate (COO–) dans les solutions alcalines. Le carboxylate est un groupe fonctionnel clé pour de nombreux dispersants de contrôle des dépôts. Le PBTC offre une bonne protection contre la corrosion, mais il est plus cher que les autres produits.
L’HPA est un ajout plus récent à la famille des tartres d’organophosphates/inhibiteurs de corrosion, et est particulièrement efficace parce qu’il forme une couche monomoléculaire avec du calcium sur les surfaces métalliques.
Les phosphonates sont presque toujours mélangés avec d’autres agents de contrôle des dépôts et des inhibiteurs de corrosion (principalement anodiques). La plage normale de contrôle des phosphonates est de 2 à 10 ppm (sous forme de PO4).
Développements de polymères
Le développement de polymères pour la modification et la séquestration des cristaux a amélioré la chimie du contrôle des dépôts. La figure 7.44 illustre divers groupes fonctionnels pour les polymères importants de traitement de l’eau.
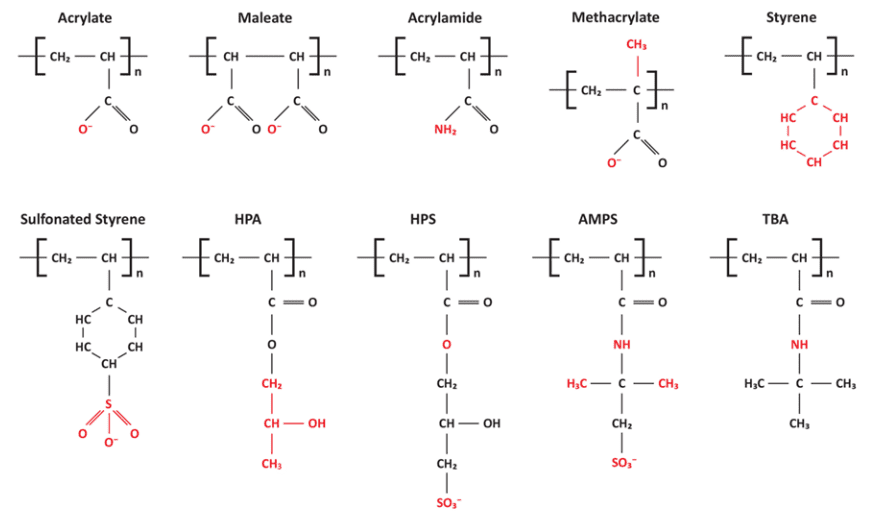
Figure 7.44. Groupes fonctionnels sur les polymères de contrôle des dépôts. HPA, acrylate d'hydroxypropyle ; HPS, 2-hydroxypropylsulfonate ; AMPS : Acide sulfonique 2-acrylamido-2-méthylpropane; TBA, acrylate de tert-butyle.
En plus des groupes fonctionnels, la structure et la taille des polymères ont une influence sur l’inhibition du tartre. Les molécules courantes sont de 500 à 15 000 daltons, mais dans certains cas, les polymères beaucoup plus gros peuvent bien fonctionner. Certains polymères ont été conçus pour contrôler le carbonate de calcium, le sulfate de calcium et les dépôts liés au fer, et d’autres pour contrôler les phosphates de calcium qui peuvent émerger des programmes de traitement par phosphate/phosphonate.
Les formulations avancées peuvent inclure des copolymères, des terpolymères et des quadruples polymères qui ont plusieurs groupes fonctionnels différents pour traiter les eaux complexes. Les composés inhibent la formation de tartre par plusieurs mécanismes, y compris la séquestration, la modification du cristal et la dispersion du cristal.
Séquestration
Comme on peut l’observer à la figure 7.44, certains composés ont les mêmes groupes fonctionnels, c.-à-d. le sulfonate et le carboxylate, que ceux des résines échangeuses d’ions pour le traitement de l’eau d’appoint (voir le chapitre 3). Pour les deux applications, les sites actifs à charge négative lient les cations, y compris le calcium et le magnésium. La principale différence est que les résines échangeuses d’ions solides sont contenues dans un réservoir tandis que les polymères solubles et mobiles de contrôle des dépôts se déplacent dans le système d’eau de refroidissement. Les noms génériques ou commerciaux courants pour ces polymères comprennent :
- Polyacrylate (PAA)
- Polyméthacrylate (PMAA)
- Polymaléate (PMA)
- Copolymère AA/ampères
Modification du cristal
Certains polymères modifient ou déforment les cristaux nés.

Les cristaux déformés ne présentent aucun des cristaux ressemblant à une aiguille ou à face plate illustrés à la Figure 7.45a, mais la structure est beaucoup plus fragile, friable et ne forme pas de gros grains de cristal. Le PA, le PMA et des composés similaires sont efficaces pour contrôler le carbonate de calcium.
Le contrôle du tartre de phosphate de calcium est plus difficile, surtout sur les surfaces de transfert de chaleur. La prévention du tartre peut nécessiter des copolymères ou des terpolymères qui comprennent des groupes de sulfonate.
Le fer présente un défi pour la chimie des polymères, car le fer se lie fortement aux sites carboxylique et sulfonate, réduisant ainsi leur efficacité pour la séquestration du calcium. Dans de nombreuses eaux, cependant, la concentration en fer est faible et ne présente pas de problèmes importants.
Dispersion des cristaux
Les dispersants polymères sont principalement chargés négativement. Les particules en suspension ont généralement une charge négative globale. Les polymères améliorent la charge négative, ce qui entraîne une répulsion accrue qui maintient les particules en suspension. La dispersion peut être efficace sur les solides en suspension de taille fine comme les produits de limon, d’argile et de corrosion, et possiblement certains débris microbiologiques. L’APA et l’AMP sont de bons produits pour la dispersion.
Un facteur souvent important pour le contrôle des dépôts est d’améliorer la capacité des polymères à pénétrer les dépôts. Cela est particulièrement vrai pour les matières organiques, y compris les huiles et les graisses, car ces composés lient les dépôts ensemble. Le biofilm est également un agent de liaison particulièrement puissant. Les surfactants peuvent aider à décomposer ces matériaux. Des composés cationiques, anioniques et non ioniques sont tous disponibles.
Les surfactants non ioniques sont similaires aux détergents en ayant un groupe fonctionnel hydrophile (amoureux de l’eau) et une chaîne lipophile (amoureux de l’huile). Lorsque l’extrémité lipophile se lie aux huiles, l’extrémité hydrophile se fixe aux molécules d’eau pour éliminer l’huile. Les modifications structurelles des sites actifs lipophiles et hydrophobes permettent une chimie de solvation spécialisée.
Les surfactants anioniques servent à la dispersion du limon et des solides en suspension. Les surfactants anioniques produisent parfois de la mousse, ce qui n’est généralement pas un problème avec les composés non ioniques.
Les dispersants cationiques sont principalement des biodispersants ou des biocides. Vous trouverez plus de détails sur ces produits chimiques dans la section Contrôle microbiologique de ce chapitre.
Méthodes de conception pour aider à contrôler le dépôt
Lorsque l’encrassement du limon ou du macrosalissure a un impact sur la performance de l’échangeur de chaleur, l’installation de l’équipement de lavage à contre-courant peut être bénéfique, si l’unité peut se détacher périodiquement pour le nettoyage.
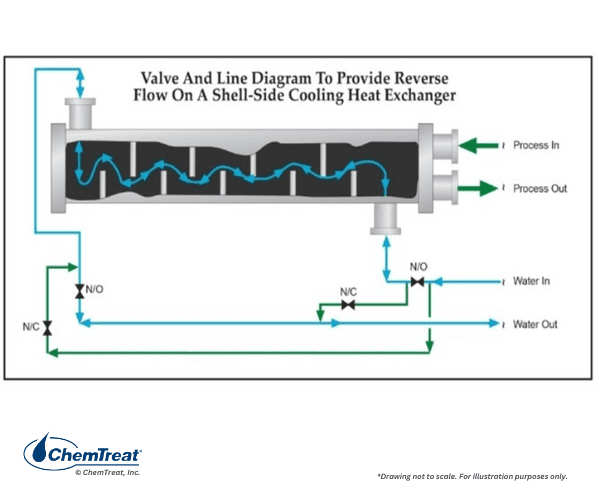
Une autre modification consiste à installer un collecteur sous l’échangeur de chaleur pour que l’air heurte l’eau côté coquille. Ceci est illustré dans le dessin ci-dessous.

Contrôle de la corrosion
Les sections d’ouverture de ce chapitre ont décrit plusieurs des problèmes de corrosion les plus importants du système de refroidissement. Nous allons maintenant examiner les techniques modernes de contrôle de la corrosion.
Lorsque les programmes d’eau de refroidissement à recirculation ouverte sont passés du traitement acide-chromate au traitement phosphate-phosphonate-polymère-zinc, une grande partie de la chimie de ces derniers programmes a également servi au contrôle de la corrosion.
Considérez encore une fois le schéma de base du mécanisme de corrosion le plus courant dans les systèmes d’eau de refroidissement, l’attaque de l’acier au carbone par l’oxygène dissous. À titre de rappel, l’oxydation et la perte du métal se produisent aux anodes, avec le transfert d’électrons et la réduction des espèces dissoutes aux cathodes.
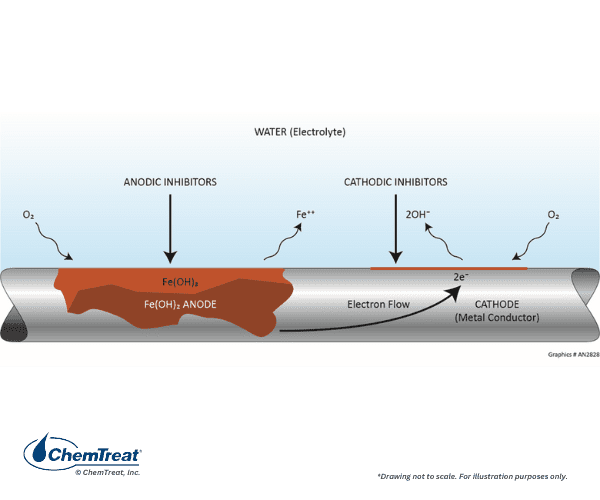
Les inhibiteurs de corrosion dépolarisent efficacement (réduisent ou arrêtent le flux électrique) la réaction de corrosion soit à l’anode ou à la cathode, soit les deux avec des programmes d’inhibiteurs mixtes. En général, les inhibiteurs cathodiques précipitent au site cathodique à pH élevé local pour former une barrière qui limite le taux de réduction de l’oxygène. Les inhibiteurs anodiques favorisent généralement la formation d’un oxyde métallique stable à la surface de l’anode. Cela limite la dissolution du métal. Un progrès considérable s’est produit avec le développement de produits filmogènes qui protègent toute la surface métallique. L’examen de cette technologie de tournage suit la discussion ci-dessous sur l’inhibition anodique et cathodique traditionnelle.
Tableau 7-3. Inhibiteurs de corrosion courants
| Anodique | Cathodique | Filmer |
| Molybdate | Phosphates organiques | Azoles (pour les alliages de cuivre) |
| Nitrite | Orthophosphate | Amines de tournage |
| Orthophosphate | Polyphosphates | Polysilicates |
| Zinc | RPSI* |
Carbonate de calcium – Inhibiteur naturel de la corrosion
La section précédente sur la formation de tartre suggère que le carbonate de calcium est le dépôt le plus naturel et souvent le plus problématique. Cependant, lorsque les concentrations d’alcalinité du calcium et du bicarbonate existent avec modération, la présence des deux peut être bénéfique. Si l’eau contient au moins 50 ppm de dureté calcique et 50 ppm d’alcalinité (les deux sous forme de CaCO3), les constituants offrent potentiellement une certaine protection contre la corrosion en tant qu’inhibiteur cathodique. La clé est qu’aux cathodes illustrées à la Figure 7.47, la production d’ions hydroxyles génère une région localisée de pH élevé. Cela peut induire la formation d’une légère couche de carbonate de calcium qui inhibe le transfert d’électrons aux cathodes.
Orthophosphate
Comme il a été décrit, l’orthophosphate est un ingrédient principal des programmes phosphate-phosphonate pour élever le pH dans une plage légèrement alcaline et minimiser la corrosion générale. De plus, l’orthophosphate réagit avec le fer (Fe2+) généré aux anodes pour former un précipité fer-phosphate qui se dépose sur les anodes et aide à inhiber les réactions électrochimiques. Les analyses de spectroscopie électronique ont montré que la couche d’inhibiteur réelle est un complexe de fer gamma avec une formule de FeOOH•FePO4. Le composé passive les anodes et étouffe les réactions de corrosion. Ce film monomoléculaire se décompose au fil du temps et nécessite donc une concentration continue de phosphate.
L’orthophosphate agit également comme inhibiteur cathodique dans certaines conditions. Tout comme le pH plus élevé localisé au niveau de la cathode précipite le carbonate de calcium et, comme nous le verrons, l’hydroxyde de zinc, il peut également précipiter le phosphate de calcium.
Polyphosphate
Comme nous l’avons mentionné, les polyphosphates séquestrent des cations multivalents comme le calcium et le fer pour inhiber la desquamation. Ces complexes développent une charge positive nette et migrent vers les cathodes pour former un dépôt de barrière. Le dépôt bloque l’oxygène de la surface, réduisant ainsi le courant de corrosion.
Zinc
Le zinc a été un additif standard pour les programmes phosphate-phosphonate, avec une concentration recommandée commune de 0,5 à 1,0 ppm. Le zinc réagit avec les ions hydroxyles produits aux cathodes pour former un précipité d’hydroxyde de zinc (Zn(OH)2) qui dépolarise les réactions cathodiques. Le zinc peut être efficace contre la corrosion par piqûres. Il a été courant de combiner le zinc avec un inhibiteur de corrosion anodique comme l’orthophosphate pour une protection complète contre la corrosion.
De nombreux composés de zinc sont très insolubles, y compris le phosphate de zinc, et, si des contaminants de sulfure sont présents, le sulfure de zinc.
Préoccupations environnementales liées à la chimie du phosphate, du phosphate et du zinc
La décharge de phosphore dans les plans d’eau naturels est une préoccupation considérable et croissante, et les effets de cette décharge sur la prolifération des algues toxiques.

À de nombreux endroits maintenant, la décharge de phosphore est limitée, voire entièrement interdite. La décharge de métaux, y compris le zinc et le cuivre, est également restreinte. Ces restrictions sont un facteur majeur dans le passage des programmes à base de phosphore aux programmes alternatifs de formation de films.

RÉDUCTION DE L’UTILISATION DE PHOSPHATE DANS UNE USINE D’AMMONIAC DOTÉE DE LA TECHNOLOGIE DE REFROIDISSEMENT FLEXPRO®
Chimie de tournage
Comme les sections précédentes l’ont suggéré, la fonction clé des inhibiteurs de corrosion est de protéger les surfaces métalliques. Les anciens programmes acido-chromatiques étaient excellents à cet égard, car au fil du temps, le chromate réagirait avec toute la surface métallique et établirait la continuité, tant que la concentration résiduelle était maintenue dans l’eau de refroidissement. Mais le changement apporté aux programmes phosphate-phosphonate a modifié cette méthodologie. L’inhibition de la corrosion est en grande partie accomplie par la précipitation de produits solides aux cathodes et anodes. Ces dépôts peuvent être nettoyés, ce qui permet une corrosion localisée. À l’inverse, la suralimentation peut induire une forte précipitation du phosphate de calcium et parfois des phosphonates de calcium.
Par conséquent, selon un effort de recherche pluriannuel discuté dans la référence 3, ChemTreat a développé une suite de programmes de contrôle de la corrosion sans phosphate/sans zinc et sans encrassement, qui « interagissent directement avec les surfaces métalliques pour former un complexe réactif d’inhibiteur de l’amidon polyhydroxylé (RPSI) qui est indépendant du calcium, du pH ou d’autres constituants chimiques de l’eau ». La chimie établit une couche protectrice directe sur les surfaces métalliques, contrairement aux programmes de phosphate/phosphonate qui reposent sur le dépôt de produits de réaction pour former des barrières protectrices, qui, comme nous l’avons noté, peuvent être difficiles à contrôler.
Un exemple classique de la nécessité d’améliorer les méthodes de protection contre la corrosion est illustré dans l’illustration suivante d’un échangeur de chaleur à tube et à coquille à deux passages, dont l’eau de refroidissement à l’époque a été traitée avec un programme traditionnel de phosphate-phosphonate.

À l’entrée de l’échangeur de chaleur (les tubes inférieurs de cet appareil), la corrosion était problématique. Du côté de la sortie du réchauffeur (la moitié supérieure), le dépôt et la formation de tartre étaient gênants. Par conséquent, le programme original n’a pas été efficace pour atténuer la corrosion ou les dépôts selon l’emplacement. Un passage à la chimie RPSI a éliminé les deux problèmes.

Contrôle de la corrosion de l’eau de refroidissement fermée (CCW)
Le refroidissement primaire dans de nombreuses industries est un aspect critique de l’exploitation, et les perturbations peuvent coûter beaucoup d’argent en perte d’efficacité et de production. Mais souvent négligés sont les systèmes auxiliaires d’eau de refroidissement fermée, qui servent également des processus vitaux. La défaillance d’un système fermé a le potentiel d’arrêter une partie, sinon la totalité, de l’usine.
Le contrôle strict de la chimie est un aspect qui rend le refroidissement fermé efficace pour de nombreuses applications. Cependant, la négligence du traitement et de la surveillance de l’eau peut entraîner la corrosion et l’encrassement.
Le terme « système d’eau de refroidissement fermé » est quelque peu une erreur d’identification, car pratiquement tous les systèmes subissent des fuites ou de petites pertes qui nécessitent un maquillage. (Si une corrosion grave s’est produite, ces pertes peuvent être importantes.) Les systèmes sont souvent conçus avec un réservoir à tête pour le maquillage et pour gérer les changements de la demande. Certains réservoirs de tête sont ouverts à l’atmosphère, ce qui permet à l’oxygène de pénétrer dans l’eau de refroidissement et d’influencer le potentiel de corrosion.
Bien qu’il puisse être possible d’utiliser de l’eau avec des qualités variées dans les systèmes CCW, un choix commun et l’objectif principal de cette discussion est le condensat ou l’eau déminéralisée qui est spécialement traitée. Le choix du condensat sur une eau moins pure minimise la possibilité de difficultés causées par les composés de dureté formant le tartre ou les agents corrosifs tels que le chlorure et le sulfate. Pour les systèmes qui pourraient potentiellement geler par temps froid, une solution de glycol peut être nécessaire. Cette chimie peut évidemment influencer les exigences de surveillance et de dosage.
Le matériau de tuyauterie typique pour les systèmes CCW est l’acier au carbone, avec l’acier inoxydable ou peut-être les alliages de cuivre étant un choix commun pour les tubes d’échangeur de chaleur ou les plaques dans un échangeur de plaque et de cadre. D’autres métaux peuvent inclure l’aluminium ou les métaux contenus dans les raccords soudés dans les serpentins de refroidissement de l’échangeur de chaleur. Lors de la planification d’un programme de traitement, il est important de connaître la métallurgie complète du système.
Les deux inhibiteurs de corrosion courants de l’eau de refroidissement sont le nitrite et le molybdate, comme indiqué ci-dessous.
Nitrite
Le nitrite (NO2‒), habituellement alimenté en nitrite de sodium (NaNO2), est un inhibiteur anodique en raison de la réaction chimique avec l’hydroxyde de fer aux anodes. Le nitrite de sodium est un produit chimique peu coûteux et sécuritaire à manipuler. Comme le montrent les réactions suivantes, le nitrite favorise la formation d’une couche passive d’oxyde de fer sur la surface métallique.
- 9Fe(OH)2 + NO2 → 3Fe3O4 + NH4 + 2OH + 6H2O | Éq. 7-24
- 9Fe(OH)2 + NO2 → 3(Fe2O3) + NH4 + 2OH + 3H2O | Éq. 7-25
L’ajout d’un agent de construction de l’alcalinité/de tamponnage pour maintenir le pH dans une plage légèrement alcaline est courant.
Les inhibiteurs anodiques comme les nitrites sont également appelés inhibiteurs « dangereux », car si les résidus tombent sous les limites du seuil, un petit nombre d’anodes se développeront dans un grand environnement cathodique. Des piqûres rapides peuvent se produire. Par conséquent, une plage de résidus de nitrite recommandée courante est de 500 à 1 000 ppm pour inhiber la corrosion générale et la corrosion par piqûre. Cependant, lorsqu’ils sont utilisés avec d’autres inhibiteurs comme le molybdate, les résidus de nitrite plus faibles peuvent être satisfaisants. Le nitrite est un excellent nutriment pour les bactéries. Nitrobactera agilis peut croître rapidement en convertissant le nitrite en nitrate. Un exemple classique provient d’une usine d’assemblage automobile, où les bactéries nitrifiantes ont bouché les petits tubes d’eau de refroidissement serpentin dans les soudeuses automatiques. Les biocides oxydants ne conviennent pas au contrôle microbiologique, car les oxydants convertissent le nitrite en nitrate. Un biocide non oxydant (voir la discussion plus loin dans ce chapitre) peut fournir un contrôle efficace.
Molybdate
Le molybdate de sodium (Na2MoO4) a été utilisé pour la première fois comme inhibiteur de corrosion en 1939 pour les systèmes de refroidissement automobiles. Il semble que le molybdate agit de la même manière que le chromate et s’adsorbe sur la matrice d’oxyde de fer aux anodes.
- Fe2+ + MoO42- → FeMoO4↓ | Éq. 7-26
Cette couche peut ensuite évoluer davantage vers ce qu’on appelle un complexe de fer gamma. Les recherches montrent que le molybdate agit comme un inhibiteur de piqûres en raison de sa capacité à s’accumuler dans la partie acide d’une fosse et à bloquer le processus de corrosion. Une plage de contrôle commune pour le molybdate est d’environ 1/3 de nitrite. Bien que le molybdate soit un oxyanion, il nécessite un peu d’oxygène résiduel pour être efficace. Une quantité suffisante d’oxygène dissous peut pénétrer dans l’eau de refroidissement ou le réservoir de la tête pour fournir la quantité nécessaire.
Le molybdate est un produit chimique coûteux, et les coûts peuvent être prohibitifs dans certaines applications.
Inhibition de la corrosion des alliages de cuivre
Le cuivre est un métal superbe pour le transfert de chaleur, et par conséquent, des alliages de cuivre ont été sélectionnés pour de nombreux tubes d’échangeur de chaleur. Bien que le cuivre soit un métal plus noble que le fer, une corrosion importante est possible dans certains environnements. Comme indiqué précédemment, la combinaison d’oxygène dissous et d’ammoniac peut être particulièrement corrosive. Les méthodes de contrôle de la corrosion les plus populaires depuis des années ont utilisé des azoles, dans un autre exemple de chimie de formation de film. La figure 7.52 illustre l’effet général.
Les azoles se lient aux atomes de cuivre sur la surface métallique par l’intermédiaire d’un groupe azote actif. L’anneau organique semblable à une plaque forme ensuite une barrière pour protéger le métal du fluide en vrac. Les dérivés azolés les plus courants sont énumérés ci-dessous.
Benzotriazole
Le 1,2,3-benzotriazole (BZT – C6H5N3) est le composé illustré à la figure 7.52. C’est l’azole le plus fondamental, mais il offre une bonne inhibition de la corrosion dans les systèmes d’eau de refroidissement en circulation.
Tolyltriazole
Le tolyltriazole (TTA – C7H7N3) est similaire au BZT, mais avec un groupe méthyle ajouté à l’anneau organique.
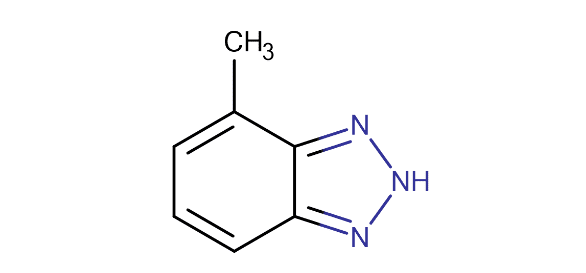
Le groupe méthyle aide à orienter la molécule afin d’établir un film barrière plus uniforme.
Un autre des premiers dérivés azolés est le 2-mercaptobenzothiazole (MBT), qui a deux groupes de soufre dans l’anneau azoté. L’un des atomes de soufre se lie également au cuivre pour former une épaisse pellicule passive.
Une difficulté avec ces composés originaux est l’attaque par les biocides oxydants, qui, bien sûr, sont nécessaires au contrôle microbiologique. Les entreprises de traitement de l’eau ont développé des dérivés azolés résistants aux halogènes qui contiennent des groupes latéraux supplémentaires pour résister à l’attaque par oxydant.
Les concentrations recommandées d’azole sont généralement maintenues dans une plage de 1 à 10 ppm et souvent de 2 à 5 ppm.
En plus de se lier à la surface métallique, les dérivés azolés forment également des complexes avec des ions de cuivre libres dans la solution. Ainsi, le cuivre dissous contribue à la « demande » d’azole, qui doit être satisfaite avant que le filmage de surface ne puisse se produire. Cependant, si l’azole est appliqué correctement, tout cuivre libre devrait rapidement disparaître et ne pas être normalement présent par la suite. Azoles demeure une partie importante de nombreux programmes FlexPro.
Amines
L’agent réducteur organique diéthylhydroxylamine (DEHA, (C2H5)2NOH)) est un passivateur métallique ainsi qu’un éliminateur d’oxygène et peut être utilisé dans des systèmes fermés. La plupart des autres agents réducteurs, y compris l’hydrazine, le carbohydrazide, l’érythorbate et le méthyléthylcétoxime, sont rarement utilisés à l’extérieur des systèmes de chaudière.
La réaction de passivation du fer par le DEHA est la suivante :
- 27Fe2O3 + (C2H5)2NOH → 18Fe3O4 + N2↑ + 4CH3COOH + 3H2O | Éq. 7-27
Les avantages des passivateurs organiques comprennent qu’ils contribuent très peu à la conductivité de l’eau, réduisent chimiquement l’oxygène, tamponnent l’eau et passivent le métal. Cela peut être très important pour certains processus de refroidissement qui ont un flux de chaleur extrêmement élevé et des températures de peau métalliques. Dans la fabrication de l’acier, par exemple, la hotte de fournaise, la lance et les électrodes sont exposées à des températures supérieures à 1 650 °C (3 000 °F). D’autres processus industriels exposent l’équipement de refroidissement à des courants électriques très élevés, comme les soudeurs automatisés dans les usines d’assemblage automobile. Une eau TDS élevée peut causer des arcs électriques et des courts-circuits qui endommagent l’équipement. Une eau de refroidissement à faible teneur en TDS (≤ 500 μS/cm) est souvent requise dans de telles applications, une exigence qui peut être possible avec les passivateurs organiques.
Inhibiteurs supplémentaires
Certains composés supplémentaires peuvent, dans certaines situations, servir d’inhibiteurs de corrosion. Les sections suivantes examinent deux d’entre elles.
Silicates
D’abord utilisé en 1921, l’orthosilicate de sodium, ou comme anion SiO32, est un inhibiteur de corrosion anodique de type précipitation semblable à l’orthophosphate. Les silicates ont été des inhibiteurs populaires dans les systèmes d’eau potable, car ils sont non toxiques et peuvent parfois être utilisés pour les applications d’eau de refroidissement.
L’orthosilicate est techniquement un mélange de monomères de silice qui coexistent avec la silice polymérisée, c’est-à-dire le polysilicate (Figure 7.54). Le pourcentage de poly vs composés orthodépendant du produit de silicate d’origine, de la concentration du silicate dans l’eau, du pH et du temps de conservation.
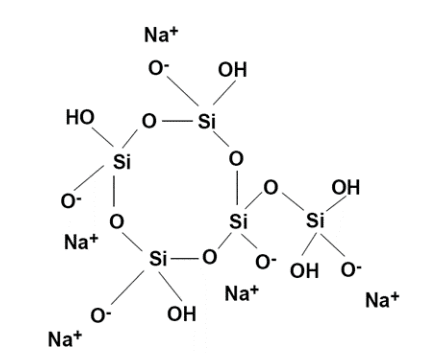
L’orthosilicate est initialement adsorbé sur les anodes à la surface du métal, où il réagit avec le fer pour former une mince couche monomoléculaire.
- Fe2+ + SiO32- + 2H2O → FeO∙Si(OH)3↓ + ½H2↑ | Éq. 7 à 28
La couche bloque le contact oxygène-métal et réduit la corrosion. Éventuellement, toute la surface métallique devient recouverte, isolant ainsi efficacement le métal des réactions électrochimiques. L’examen microscopique et radiographique de la barrière de silicate indique la formation de deux couches, la majeure partie de la silice dans une couche de surface adjacente à l’eau.
Une ligne directrice courante est une dose initiale de silicate pour établir un résidu de 25 ppm au-dessus de la concentration de fond. 30 à 60 jours de fonctionnement à ce niveau est typique. La concentration peut ensuite être réduite à 8 à 12 ppm au-dessus de l’arrière-plan pour un contrôle normal. Le film ne s’accumule pas sur lui-même et ne formera donc pas de tartre. Cependant, le traitement peut être très problématique dans les systèmes à recirculation ouverte ou les échangeurs de chaleur avec transfert de chaleur élevé en raison du potentiel de formation de tartre de silicate de magnésium ou de calcium. Un autre agent encrassant potentiel est le silicate d’aluminium, qui pourrait en résulter si l’alun est utilisé comme coagulant pour la clarification préalable au traitement de l’eau d’appoint.
Borate
Le tétraborate de sodium pentahydraté (Na2B4O7•5H2O) est un inhibiteur anodique qui protège les métaux ferreux de la corrosion. Il semble que le borate produit une couche de borate ferrique sur les surfaces métalliques, encourageant la formation d’oxyde ferrique qui agit comme une barrière au transport des ions ferriques, en particulier les ions ferriques, à partir de la surface métallique. Étant un inhibiteur relativement faible, le borate complète d’autres inhibiteurs comme le nitrite ou le silicate. Une caractéristique clé des borates est une bonne capacité de tamponnement à un pH légèrement basique de 8,3 ou légèrement supérieur.
Les borates et l’acide borique peuvent protéger contre diverses formes de dégradation du bois par les champignons, mais les concentrations nécessaires sont beaucoup plus élevées que celles qui peuvent être raisonnablement maintenues dans les systèmes de refroidissement à recirculation.
Contrôle de la corrosion par sélection des matériaux et électrochimie
Dans certaines applications, des méthodes sont disponibles pour protéger les métaux par des méthodes mécaniques ou électriques. Cette section examine l’une de chacune, galvanisant du côté des matériaux et l’utilisation d’anodes sacrificielles d’un aspect électrique.
Galvanisation
La galvanisation de l’acier au carbone est courante pour de nombreuses applications, y compris les composants structurels de la tour de refroidissement. Les petites tours de refroidissement galvanisées (voir Figure 6.9 dans le chapitre précédent) peuvent souvent être idéales d’un point de vue de l’équilibre entre la stabilité structurelle et le coût.
Le conditionnement approprié du revêtement galvanisé est essentiel à la longue durée de vie du zinc pour établir une patine protectrice en zinc gris foncé. Cela peut être réalisé en exposant les composants galvanisés à l’atmosphère pendant plusieurs mois. Comme le montre la figure 7.28, les garde-corps le long des routes et autres structures en zinc exposées à l’atmosphère développent cette patine naturellement.
Pour les composants de la tour de refroidissement qui sont installés sans exposition atmosphérique à long terme, le conditionnement du matériau pendant peut-être deux mois avant que l’unité ne soit mise en service est nécessaire pour éviter la formation de « rouille blanche ». Les détails de cette chimie sont décrits plus loin dans la section « Nettoyage pré-opérationnel et passivation » de ce chapitre.
Protection électrique : Anodes de courant sacrificielles ou imprimées
La technologie d’anode sacrificielle est disponible depuis des décennies pour protéger les tuyaux, les réservoirs, les boîtes à eau du condenseur et d’autres équipements. Dans ces applications, un métal plus réactif que l’acier, p. ex., magnésium, aluminium, zinc, est couplé à la structure dans des emplacements stratégiques. Lorsque des cellules de corrosion se développent, les électrons s’écoulent de l’élément le plus réactif à travers les fils de connexion à la structure et aux sites cathodiques sur l’acier. Cela laisse l’acier intact au détriment des blocs de métal facilement remplaçables.
Selon la référence 4, une version plus avancée du processus est impressionnée par la protection cathodique actuelle dans laquelle « une source externe d’alimentation à courant continu est connectée (ou impressionnée) entre la structure et les anodes du lit de mise à la terre ». Ces systèmes sont courants sur les pipelines. Étant donné que le courant est généré directement, les anodes moins réactives que le magnésium et l’aluminium sont acceptables et peuvent inclure de l’acier à faible teneur en alliage.
Les systèmes d’anodes actuelles sacrificielles et impressionnées doivent être soigneusement conçus, car ils peuvent être sous-spécifiés ou surspécifiés. Un courant trop faible permettra au métal primaire de se corroder aux anodes localisées, tandis qu’un courant trop élevé peut générer des réactions à la surface du métal qui causent une attaque, par exemple, un fragilisation à l’hydrogène. Une discussion plus approfondie dépasse la portée de ce livre.
Préoccupations microbiologiques
Les systèmes d’eau de refroidissement offrent un environnement idéal, chaud et humide, pour la croissance microbiologique. Les bactéries peuvent former des colonies dans de nombreux endroits, les champignons attaqueront le bois de la tour de refroidissement et les algues proliféreront dans les zones ensoleillées, en particulier sur les ponts et autres zones exposées. Les micro-organismes plus avancés tels que l’amoeba et les protozoaires se multiplient souvent dans les colonies microbiennes établies. Ces organismes plus complexes peuvent améliorer la croissance de la bactérie Legionella.
Pour contrôler les micro-organismes, les biocides homologués par l’EPA, à la fois oxydants et non oxydants, sont couramment utilisés, parfois en association avec un biopénétrant/biodispersant. Cette section traite des microbes courants qui prospèrent dans les systèmes d’eau industriels et décrit les stratégies de contrôle.
Bactéries
Les bactéries sont constituées de procaryotes unicellulaires (microbes qui n’ont pas de noyau cellulaire défini) et peuvent être aussi petites que 0,1 micron. Les bactéries peuvent être classées en cinq groupes selon leurs formes de base : sphérique (coques), tige (bacilles), spirale (spirille), virgule (vibrios) ou tire-bouchon (spirochaètes). Elles peuvent exister sous forme de cellules uniques, en paires, en chaînes ou en grappes. Cependant, la forme seule n’est pas un bon indicateur d’un type bactérien, car une seule souche de bactéries peut prendre différentes formes selon les conditions de croissance. Différentes espèces peuvent avoir des morphologies similaires.
Les bactéries ont un large éventail de besoins en nutriments, en oxygène et en énergie. Les bactéries aérobies nécessitent de l’oxygène, tandis que les bactéries anaérobies prospèrent sans oxygène. Les anaérobies facultatifs sont des microbes qui produisent de l’énergie par respiration aérobique si de l’oxygène est présent, mais qui peuvent passer à un processus de fermentation si de l’oxygène est absent. Les bactéries sont catégorisées comme étant soit planctoniques (flottantes libres) dans l’eau en vrac, soit sessiles dans les colonies fixées aux surfaces. Les microbes sésiles sont responsables des biofilms et sont généralement la principale préoccupation en matière d’impact sur la performance du système d’eau.
Les bactéries sont également regroupées par plages de température optimales pour la croissance; bactéries thermophiles, au-dessus de 50 °C (122 °F), mésophiles dans une plage de 20 à 37 °C (68 à 99 °F) et psychrophiles à 4 à 10 °C (39 à 50 °F). Les microbes qui peuvent croître et prospérer dans des environnements extrêmes sont appelés extrémophiles, et bien que chaque espèce de bactéries se développe mieux dans une plage de température optimale, la plupart peuvent également s’adapter et croître à des températures en dehors de la plage optimale.
Dans la plupart des systèmes d’eau de refroidissement industriels modernes, le pH est maintenu dans une plage alcaline pour le contrôle de la corrosion, ce qui est idéal pour la croissance bactérienne. Les bactéries les plus courantes sont celles du genre Pseudomonas, mais les autres espèces comprennent :
- Bactéries réduisant le sulfate (SRB)
- Bactéries liées au fer (CEE)
- Bactéries formant de la slime
- Bactéries nitrifiantes
- Bactéries dénitrifiantes
Lorsque des colonies se développent, des aérobies, des anaérobies et des organismes facultatifs se forment généralement dans la biomasse.
Champignons, levures et moisissures
Le terme champignons comprend les levures et les moisissures. Les champignons sont un groupe d’organismes eucaryotes non photosynthétiques dont la taille est supérieure à celle des bactéries. La plupart des champignons sont aérobies et nécessitent de l’oxygène pour survivre. Les levures, comme celles pour la bière ou la vinification, sont des anaérobies facultatifs. Les champignons sont des organismes importants pour de nombreux processus de traitement des déchets lorsqu’ils décomposent les matières organiques. Dans les systèmes industriels d’eau de refroidissement, ces attributs peuvent être nocifs. Les champignons Basidiomycetes et Ascomycetes peuvent attaquer le bois de la tour de refroidissement.
Alors que la levure est un type de champignons qui ne contient que des cellules uniques, les moisissures contiennent plusieurs noyaux identiques. Les colonies sont caractérisées par des filaments filamenteux multicellulaires (et parfois colorés) appelés hyphes. Les moisissures sont importantes sur le plan industriel pour la production d’antibiotiques et de fromage.
Les champignons prospèrent dans les environnements acides et sont moins actifs dans les systèmes d’eau de refroidissement modernes par opération typique dans une plage de pH légèrement basique. Cependant, il n’est pas rare de trouver des champignons dans les dépôts de biofilm, ce qui suggère un environnement acide dans le biofilm.

Algues
Les algues sont un groupe diversifié d’organismes aquatiques photosynthétiques qui nécessitent la lumière du soleil. Les algues sont des photoautotrophes, ce qui signifie qu’elles peuvent produire de nombreux nutriments nécessaires à partir du dioxyde de carbone, de l’eau et de la lumière du soleil. Le phosphate est un nutriment essentiel supplémentaire et un facteur limitant la croissance.
Dans les systèmes de refroidissement à recirculation ouverte, les algues poussent dans les zones exposées au soleil, notamment les ponts des tours à circulation transversale. Bon nombre de ces tours ont couvert des terrasses pour bloquer la lumière du soleil, limitant ainsi les possibilités de croissance des algues. Sans vérification, l’encrassement algal peut entraîner un débit d’eau déséquilibré dans la tour de refroidissement et une efficacité de fonctionnement réduite. Les algues peuvent cependant apparaître dans d’autres zones ensoleillées.

Microbes et biofilms
Comme nous l’avons déjà mentionné, les microbes présents dans l’eau de refroidissement sont flottants ou sésiles. Les conditions du système dicteront dans une certaine mesure le développement de sessile. Par exemple, des vitesses élevées peuvent physiquement empêcher les microbes de se déposer. Lorsque les microbes se fixent aux surfaces, certains organismes commencent presque immédiatement à produire une couche protectrice de polysaccharides, communément appelée biofilm ou slime. Les aérobes prolifèrent généralement dans les couches externes du biofilm (près du liquide en vrac) tandis que les anaérobies se développent dans des zones à faible concentration d’oxygène, c.-à-d. à la base du biofilm. La boue emprisonne d’autres microorganismes, le limon, les produits de corrosion et d’autres débris dans les eaux industrielles.
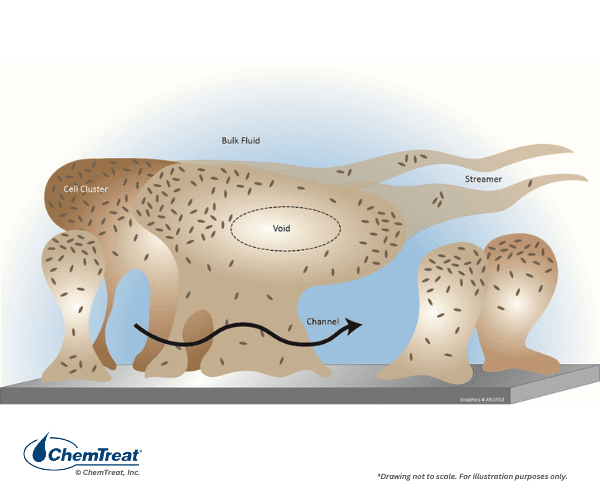
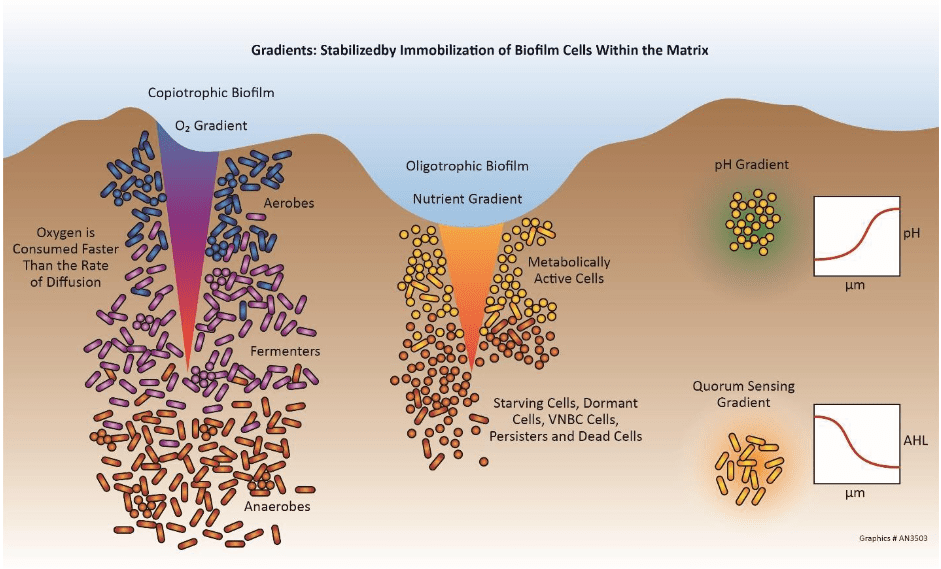

Crédit photo : CBE-MSU Bozeman.
Le biofilm protecteur rend les microorganismes sessiles plus difficiles à tuer que les microbes planctoniques. Les travaux du professeur Phil Stewart de l’Université d’État du Montana ont montré que le mouvement de toute molécule dans un biofilm est limité par la diffusion et la réactivité. Par conséquent, l’étendue de la pénétration du biocide est limitée par la réactivité de la molécule vers la slime protectrice. Nous examinerons ces concepts sous peu. Idéalement, le système d’alimentation en biocide d’eau de refroidissement/service d’une usine a été conçu et fonctionne correctement pour tuer les organismes planctoniques avant qu’ils puissent établir des colonies. Cependant, le personnel de l’usine doit souvent traiter le traitement des colonies bien établies.
Formes de vie plus élevées – Amoeba et protozoaires
Comme indiqué précédemment, si les colonies microbiologiques deviennent bien établies, des formes de vie plus élevées apparaissent souvent. L’amoeba et les protozoaires sont des microbes eucaryotes unicellulaires omniprésents. La cellule protozoaire peut avoir des appendices spécialisés comme des cils ou des flagelles pour le mouvement.
Bien que de nombreux types d’amoba puissent causer des problèmes de santé humaine, la relation entre l’amoba, les protozoaires et la bactérie Legionella est bien connue. La légionelle est l’organisme découvert pour la première fois en 1976 lorsqu’il a infecté des personnes assistant à un congrès de la Légion américaine à Philadelphie. Près de trois douzaines de personnes sont mortes et beaucoup d’autres sont tombées malades. Il a été retracé à la vapeur d’eau contenant les bactéries dans le panache d’échappement d’une tour de refroidissement sur le toit de l’hôtel du congrès. La vapeur est entrée dans l’admission d’une unité de traitement d’air et s’est propagée à travers l’hôtel.
La littérature scientifique contient un ensemble fascinant de preuves qui documentent la relation entre la Legionella et l’amoeba. Le lien entre la légionelle et l’amoba a d’abord été démontré par Rowbotham qui a montré que l’amoba pouvait servir d’hôte pour la légionelle. Peu de temps après, Kurtz a décrit l’isolement de l’amoebae dans les eaux de refroidissement qui contiennent de la légionelle, apportant ainsi un soutien supplémentaire à la théorie de Rowbotham sur la relation entre les deux. Un rapport ultérieur de Rowbotham a ensuite offert un mécanisme pour la croissance et la prolifération de la Legionella au sein de l’espèce d’Amoebae, Acanthamoeba polyphaga.
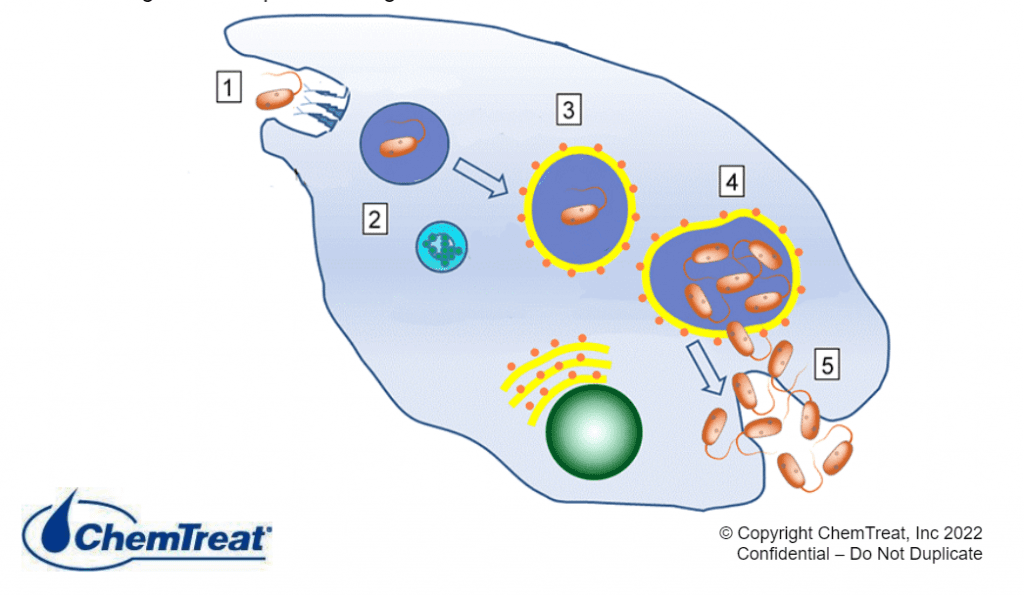
De nombreux rapports ont émergé sur les mécanismes de croissance de la Legionella dans ces organismes plus élevés et leur libération, mais le résultat est que la conception et le fonctionnement appropriés du système d’alimentation en biocide sont essentiels pour minimiser toutes les formes d’encrassement microbiologique. Une autre mesure importante est de trouver et d’éliminer toute la tuyauterie des jambes mortes. L’eau stagnante peut permettre aux organismes de proliférer.
L’amoeba et les protozoaires sont beaucoup plus résistants aux effets biocides des halogènes que les microorganismes plus simples, mais sont sensibles à plusieurs biocides non oxydants couramment utilisés.
Encrassement macroscopique
L’encrassement des surfaces par des organismes plus grands que les bactéries ou les champignons est considéré comme un macroencrassement. Les organismes courants sont les moules, les palourdes et les barnacles.
Les palourdes d’eau douce sont devenues problématiques aux États-Unis à la fin des années 1970 lorsque la palourde asiatique, Corbicula flominea, est entrée dans le pays. Vers 1986, Dreissena polymorpha, la moule zébrée, a été introduite dans les Grands Lacs. Ces moules sont originaires de la mer Noire et Caspienne et peuvent atteindre des densités allant jusqu’à 700 000 personnes par mètre carré.

Les deux organismes présentent d’importants défis en matière de consommation d’eau et de systèmes, mais les moules zébrées ont été au centre de leurs préoccupations au cours des dernières années, après leur propagation des Grands Lacs par divers cours d’eau dans l’est et le milieu-ouest des États-Unis.
Contrôle microbiologique
Comme note préliminaire à la discussion suivante sur les biocides de l’eau de refroidissement, de nombreux pays, y compris les États-Unis, le Canada et le Mexique, ont imposé des limites à l’utilisation de composés antimicrobiens dans les milieux industriels. L’enregistrement du produit est requis, ce qui comprend généralement des limitations sur la façon et l’endroit où le produit peut être appliqué. Les produits avec le même type et le même niveau de composants actifs, mais avec des noms commerciaux différents, doivent chacun être enregistrés en tant qu’entités distinctes. Aux États-Unis, les enregistrements relèvent de la compétence de l’Environmental Protection Agency (EPA) et de la Federal Insecticide, Fungicide and Rodenticide Act (FIFRA).
En plus de l’enregistrement fédéral, des règlements d’État/territoire/provinciaux supplémentaires peuvent être placés sur l’utilisation de produits antimicrobiens. La plupart des entreprises, en particulier les installations plus grandes, nécessitent également des processus finalisés avant qu’un nouveau produit chimique puisse être appliqué. Les usines doivent également respecter les limites de décharge pour certains composés. Aux États-Unis, ces règlements sont sous les auspices du National Pollution Discharge Elimination System (NPDES) créé dans le cadre de la Clean Water Act (CWA) de 1972. Les règlements exigent des permis approuvés avant qu’un flux d’eau contenant des produits chimiques de traitement, y compris des biocides, puisse être déversé dans l’environnement. Les États individuels doivent faire respecter les règlements du NPDES, mais sont libres d’imposer des directives plus strictes si désiré.

SpecOx® est le programme unique d’oxydants spécialisés de ChemTreat, qui offre un contrôle microbien amélioré dans les eaux de refroidissement et de traitement.
Biocides oxydants et non oxydants
Pour la grande majorité des eaux de refroidissement à recirculation et à passage unique, les biocides oxydants sont le pilier du programme de contrôle microbien, car la chimie représente généralement la méthode la plus rentable pour maintenir la propreté du système. Les systèmes plus difficiles à traiter peuvent également bénéficier d’un biocide non oxydant supplémentaire et/ou de l’ajout d’un biopénétrant/biodispersant. Les composés les plus courants dans chaque catégorie sont abordés ci-dessous.
Biocides oxydants
De nombreuses références suggèrent que 1893 est l’année au cours de laquelle le chlore a été appliqué pour la première fois comme biocide d’eau potable, avec un développement rapide de la technologie au début des années 1900.
Le chlore gazeux a été le bourreau de travail pour boire de l’eau et pour refroidir le traitement de l’eau pendant de nombreuses années. Les bouteilles d’une tonne sont devenues la méthode d’entreposage dans de nombreuses installations, de l’eau potable et autres. Lorsque du chlore est ajouté à l’eau, la réaction suivante se produit :
- Cl2 + H2O � rÈGLEMENT HOCl + HCl | Éq. 7-29
L’HOCl, l’acide hypochloreux, est l’agent tuant, et il fonctionne en pénétrant les parois cellulaires, puis en oxydant les composants cellulaires internes. L’efficacité et le pouvoir tuant de ce composé sont grandement influencés par le pH en raison de la nature d’équilibre du HOCl dans l’eau, comme illustré ci-dessous.
- xHOCl �. TAILLE H+ + OCl- | Éq. 7 à 30x
L’OCl- est un biocide beaucoup plus faible que l’HOCl, probablement parce que la charge sur l’OCl-ion ne lui permet pas de pénétrer efficacement les parois cellulaires. La dissociation de l’acide hypochloreux augmente considérablement par rapport à l’augmentation du pH.
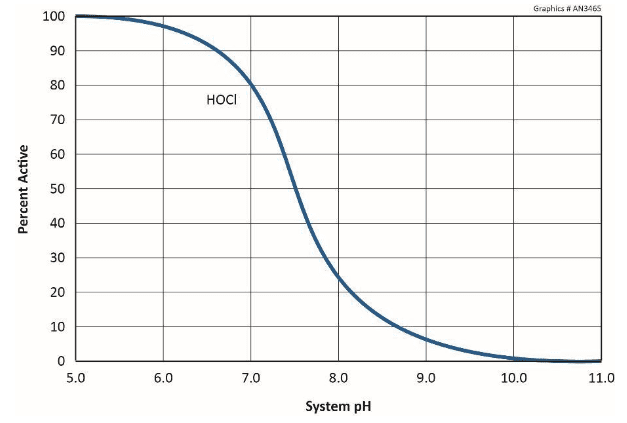
Étant donné que de nombreux programmes de traitement de tartre/corrosion de la tour de refroidissement fonctionnent à un pH alcalin, souvent près ou légèrement supérieur à 8,0, la chimie du chlore droit peut ne pas être le meilleur choix.
En raison de problèmes de sécurité liés au chlore gazeux, de nombreuses installations industrielles sont passées à l’hypochlorite de sodium liquide (NaOCl, c’est-à-dire javellisant), avec une concentration courante de chlore actif de 12,5 %. L’eau de Javel contient souvent une petite quantité d’hydroxyde de sodium, donc lorsqu’elle est injectée dans le flux d’eau de refroidissement, elle augmente le pH, peut-être seulement légèrement. Cependant, si l’eau est alcaline pour commencer, la majeure partie du réactif existera sous forme d’OCl–ion. Une solution de rechange est la production d’hypochlorite sur place, qui s’est avérée efficace dans certaines applications.
Demande de chlore
En tant qu’oxydant puissant, l’acide hypochloreux peut réagir avec des composés qui sont souvent présents dans les eaux de refroidissement et de traitement de recirculation. Les plus importants sont l’ammoniac et les matières organiques. La somme de ces réactions non antimicrobiennes est appelée « demande de chlore ». Les réactions consomment du biocide et réduisent la concentration disponible pour attaquer les microbes. Certaines réactions peuvent également produire des matières organiques halogénées, dont la concentration de décharge peut être régulée.
Alternatives au chlore
Comme nous l’avons noté, le pouvoir tueur du chlore diminue avec l’augmentation du pH. Une réponse populaire à cette difficulté a été la chimie du brome, où un oxydant de chlore (le javellisant est le choix commun) et du bromure de sodium (NaBr) sont mélangés dans un courant de glissement et injectés dans l’eau de refroidissement. La réaction produit de l’acide hypobromeux (HOBr), qui a des pouvoirs de destruction similaires à ceux du HOCl, mais fonctionne plus efficacement au pH alcalin.
- HOCl + NaBr � r r HOBr + NaCl | Éq. 7-31
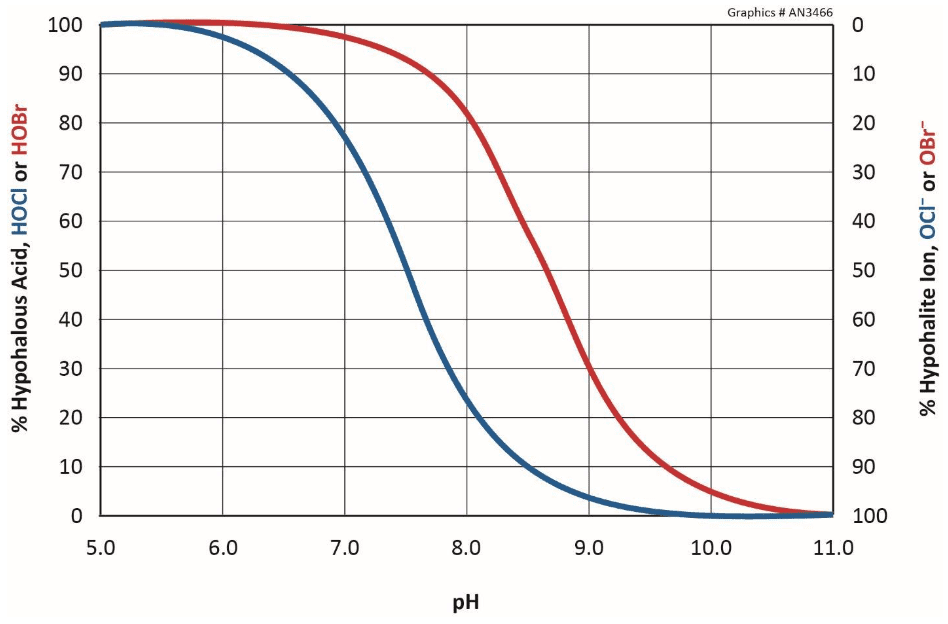
Comme l’acide hypochloreux, l’acide hypobromeux est un oxydant puissant qui a également une demande halogène. Cependant, contrairement au chlore qui réagit de façon irréversible à l’ammoniac, la réaction brome-ammoniac est réversible et laisse donc le brome libre pour l’activité contre les microbes.
Stabilisateurs halogènes
Plusieurs composés chimiques sont disponibles pour stabiliser le chlore et le brome, puis libérer les oxydants progressivement, et là où ils sont le plus nécessaires. L’halogène stabilisé présente généralement une puissance d’oxydation inférieure à celle de l’halogène parent, mais cette réduction de l’oxydation offre en fait des avantages en ce qui concerne le contrôle microbien en ce sens qu’elle réduit les réactions indésirables avec la slime protectrice.
Trois classes de stabilisateurs dominent le marché : sulfamate, diméthylhydantoïne et isocyanurates.
La structure du sulfamate est illustrée ci-dessous.
Les halogènes stabilisés par le sulfate sont peut-être les composés les plus courants de ce type. Ils sont disponibles sous forme liquide et sont principalement offerts sous forme de mélanges d’espèces de brome et de chlore.
Les espèces de chlore stabilisé et de brome diméthylhydantoïne sont une autre option.
Ces composés sont disponibles sous forme de comprimés/palettes, de granules et de poudres. Chaque produit possède des caractéristiques de dissolution individuelles, qui nécessitent une évaluation minutieuse lors de la sélection d’un système d’alimentation. Une conception commune pour l’alimentation de produits solides est un petit vaisseau dans lequel les comprimés/palettes peuvent être chargés, et qui se dissolvent ensuite graduellement dans un courant de glissement d’eau de refroidissement qui retourne au système principal. Les systèmes liquides qui produisent de l’hydantoïne stabilisée sur demande par la réaction de l’hypochlorite de sodium avec la diméthylhydantoïne liquide sont également possibles.
Les isocyanurates, disponibles principalement sous forme de solides, complètent le trio de classes halogènes stabilisées. Les formulations solides sont assez stables, ce qui permet une longue durée de conservation.
Problèmes de compatibilité
Plusieurs des mêmes limites existent pour le chlore et le brome pour les halogènes stabilisés. Les produits doivent d’abord être mélangés avec de l’eau et non pas directement avec d’autres produits chimiques qui pourraient causer une contamination ou des réactions indésirables. Les formulations de produits liquides introduisent souvent une certaine caustique dans l’eau.
Dioxyde de chlore
Le dioxyde de chlore (ClO2) est un gaz à température ambiante stable et soluble dans l’eau jusqu’à une concentration maximale d’environ 3 000 ppm. Il doit être préparé sur place par réaction de chlorite de sodium (NaClO2) ou de chlorate de sodium (NaClO3) avec un agent oxydant supplémentaire dans des conditions acides. Le dioxyde de chlore est plus cher que les halogènes, mais les techniques de production modernes ont réduit le coût.
L’état d’oxydation du chlore dans la molécule ClO2 est de +4, ce qui la rend assez réactive à de nombreux composés. Cependant, le dioxyde de chlore présente un degré élevé de sélectivité de réaction et il peut pénétrer les biofilms pour attaquer les microbes. La sélectivité est avantageuse pour d’autres applications de traitement de l’eau, y compris la destruction du phénol des eaux usées et le contrôle des odeurs.
Étant donné que le dioxyde de chlore existe en tant que gaz dans la solution, il est facilement éliminé par l’aération. Le ClO2 doit être introduit sous la surface des eaux de réception pour minimiser les pertes. La manipulation des précurseurs chimiques pour le dioxyde de chlore, qui peut inclure l’acide sulfurique, nécessite une attention particulière à la sécurité, bien que les générateurs modernes de ClO2 soient généralement conçus en pensant à la sécurité. Le strict respect des directives opérationnelles est important.
Chloramines
Les chloramines servent au contrôle microbien des systèmes d’eau industriels depuis plus d’un siècle. Dans l’eau contenant de l’ammoniac, une alimentation continue en chlore produira une série de chloramines, en commençant par la monochloramine (NH2Cl), puis la dichloramine (NHCl2) et enfin le trichlorure d’azote (NCl3). La solution atteint un « point de rupture » une fois que toute l’ammoniac a été consommé, sur lequel le chlore libre apparaît.
La monochloramine est le composé d’intérêt pour le contrôle moderne de l’encrassement biologique, et des technologies sont maintenant disponibles pour produire un flux immaculé de NH2Cl à cette fin. Comparativement à l’hypochlorite de sodium, la monochloramine est moins réactive, mais presque aussi toxique. La réactivité réduite lui permet de pénétrer les biofilms et d’attaquer les organismes sous-jacents. Cependant, la monochloramine a généralement besoin d’un temps de contact plus long que l’hypochlorite pour obtenir la destruction microbienne souhaitée.
Oxydants de peroxyde : Peroxyde d’hydrogène, acide peracétique, acide percarbonique
Plusieurs biocides/désinfectants à base de peroxyde sont sur le marché, et tous peuvent produire le radical de peroxyde (OH∙) comme composé réactif. Les produits les plus courants sont le peroxyde d’hydrogène, l’acide peracétique et l’acide percarbonique.
Contrairement au dioxyde de chlore, les peroxydes réagissent sans discrimination avec un large éventail de composés, y compris les microbes. La réaction se produit à la paroi cellulaire et les dommages provoquent la lyse cellulaire et la mort. Contrairement aux halogènes, l’oxygène est le sous-produit des réactions ou de l’autodécomposition du biocide. Un avantage secondaire est que la libération d’oxygène peut transformer l’eau de l’anaérobie à l’aérobie.
Un désavantage des biocides/désinfectants à base de peroxyde est le potentiel de désactivation des microbes producteurs de catalase. La catalase est une enzyme produite par toutes les cellules vivantes pour la protection contre le peroxyde d’hydrogène généré métaboliquement. Cependant, certains microbes produisent de grandes quantités de cette enzyme et augmenteront la production en présence de taux élevés d’H2O2. Cela rend l’utilisation à long terme du peroxyde comme désinfectant problématique. Cependant, pour un nettoyage hors ligne périodique, le peroxyde est extrêmement efficace pour éliminer les biofilms et autres dépôts organiques, en particulier lorsqu’il est utilisé en association avec un caustique, un surfactant et à des températures chaudes.
Biocides non oxydants
Bien que les produits chimiques oxydants servent normalement de base aux programmes de biocide de l’eau de refroidissement, les microorganismes peuvent développer une immunité partielle, en grande partie à partir des biofilms protecteurs. Par conséquent, dans certaines applications, l’alimentation d’un biocide non oxydant sur une base périodique, p. ex., une ou deux fois par semaine pendant environ une heure, peut aider à contrôler la croissance microbienne. Alors que les biocides oxydants endommagent généralement les parois cellulaires, les non-oxydants pénètrent les parois cellulaires pour ensuite réagir avec les composés dans la cellule qui sont nécessaires à la vie. Les sections suivantes examinent plusieurs des non-oxydants les plus courants.
Bronopol
Le bronopol est un diol halogéné qui contient un atome de carbone électrophile. Il est largement utilisé dans les applications de traitement de l’eau et, comme le 2,2-dibromo-3-nitrilopropionamide ci-dessous, a vu des applications limitées dans le champ pétrolifère. Le bronopol est particulièrement efficace contre la bactérie Pseudomonas. Le composé semble fonctionner par différents mécanismes chimiques selon que les conditions sont aérobies ou anaérobies. Le bronopol n’est pas un biocide à action rapide. Le composé peut libérer du formaldéhyde lors de la décomposition, mais le formaldéhyde n’est pas responsable de ses propriétés biocides.
Le bronopol sera hydrolysé dans des solutions aqueuses à des débits variables selon le pH. Le taux d’hydrolyse est beaucoup plus rapide à pH alcalin. L’augmentation de la température augmente le taux d’hydrolyse à un pH donné. La plage de pH optimale pour l’efficacité du bronopol est de 5 à 9. Le composé réagira avec les sulfures et les agents d’épuration d’oxygène à base de bisulfites et sera désactivé par ceux-ci. Enfin, le bronopol possède le potentiel de libérer des nitrites qui, en présence d’amines secondaires, peuvent former des nitrosoamines.
Figure 7.71. Structure de la DBNPA.
Le DBNPA est un amide halogéné largement utilisé dans le traitement de l’eau et les applications de pâtes et papiers. Dans le domaine pétrolier, la DBNPA sert à traiter l’eau d’appoint pour les fluides de fracturation. L’action biocide de la DBNPA est très rapide et les concentrations résiduelles s’hydrolysent rapidement en sous-produits moins toxiques. L’hydrolyse rapide peut être avantageuse si le flux d’eaux usées se décharge par un bassin de rétention, ce qui permet au résidu de se décomposer sans avoir besoin d’un produit chimique de désactivation. Le DBNPA s’est avéré efficace pour contrôler l’encrassement microbiologique dans les systèmes d’eau d’appoint de haute pureté, en particulier les unités d’osmose inverse.
La présence de deux atomes de brome au deuxième carbone de DBNPA rend le site très électrophile, qui réagit ensuite avec les acides aminés nucléophiles contenant du soufre méthionine et cystéine dans les cellules microbiennes. La réaction est irréversible et le résultat est l’inactivation de la protéine qui contient ces acides aminés. La mort cellulaire s’ensuit.
La plage de pH optimale pour une efficacité maximale de la DBNPA est de 4 à 8. Le taux d’hydrolyse augmente avec l’augmentation du pH et le composé perd rapidement sa puissance au-dessus de 8 pH. L’hydrolyse augmente également avec la hausse des températures. Le DBNPA est désactivé par les sulfures et les éliminateurs d’oxygène à base de bisulfites. Le composé n’est pas stable à la lumière UV et réagit avec l’ammoniac.
Glutaraldéhyde
Le glutaraldéhyde a été largement utilisé dans les applications industrielles de traitement de l’eau, y compris les opérations pétrolières et gazières, l’industrie du papier et la stérilisation des instruments médicaux. Le produit chimique réagit avec deux des vingt acides aminés essentiels, la lysine et l’arginine, nécessaires à la fonction cellulaire. L’efficacité est optimale dans une plage de pH de 7 à 10, mais le composé est plus stable à un pH acide. Le glutaraldéhyde réagira avec les éliminateurs d’oxygène à base de bisulfite pour former un produit d’ajout d’aldéhyde-bisulfite. Cette réaction est réversible et à des températures élevées (> 60 °C), le complexe se décomposera pour libérer le glutaraldéhyde. La réaction du glutaraldéhyde avec des amines ou des ions d’ammonium n’est pas réversible et produit un sous-produit avec peu ou pas d’efficacité biocide.
Isothiazolones
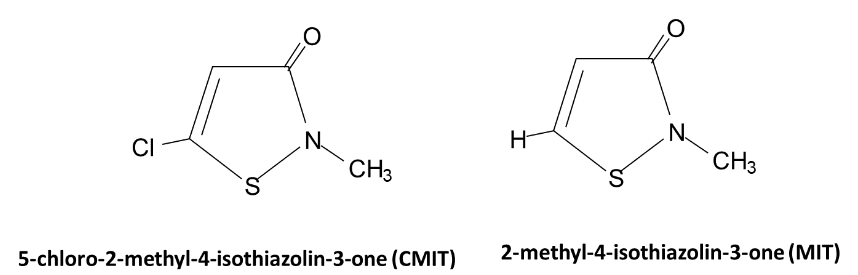
En plus des applications d’eau de refroidissement, les formulations d’isothiazoline sont, en très faibles concentrations, un agent antimicrobien courant dans de nombreux détergents. La formulation biocide à base d’isothiazolone la plus courante pour le traitement de l’eau de refroidissement contient un mélange 3 :1 de CMIT et de MIT. Les concentrations du CMIT et du MIT dans les produits enregistrés sont soit 1,5 pour cent ou 4 pour cent d’ingrédient actif. Divers stabilisateurs font partie des formations industrielles, y compris le nitrate de cuivre, le nitrate de magnésium et l’iode de potassium. Un produit actif à 1,5 % stabilisé avec du bronopol est également disponible. La sélection appropriée du produit est souvent influencée par le stabilisateur. Les isothiazolones sont des bactéricides à large spectre, mais à action lente qui montrent également une bonne réactivité contre les champignons.
Le CMIT et le MIT sont incompatibles avec le sulfure d’hydrogène ou d’autres nucléophiles à base de soufre. Par conséquent, si des bactéries réduisant le soufre (SRB) sont présentes, les isothiazolones peuvent ne pas être le meilleur choix non oxydant. Un pH élevé (> 9,5) réduira le CMIT, mais le MIT est stable à un pH supérieur à 10. Les isothiazolones peuvent être désactivés avec du bisulfite de sodium à un rapport de 2 :1 mole par rapport à l’isothiazolone.
Amines quaternaires
Les amines quaternaires, ou « quats » comme on les appelle communément, ont été largement utilisées dans une grande variété d’applications d’eau de refroidissement et de traitement. Les molécules sont chargées positivement, avec quatre groupes alkyles attachés à un atome d’azote central. Un ou plusieurs des groupes alkyles sont constitués d’une chaîne de méthyle, de benzyle, de décyl (C10), de coco (C14) ou de soya (C18). Les quaternaires sont généralement alimentés en association avec d’autres biocides et sont généralement très rentables. Certains composés servent également d’inhibiteurs de la corrosion par filmage-amine.
Les quaternaires ont des propriétés surfactantes qui solubilisent les membranes cellulaires, entraînant des dommages cellulaires et la mort. Les composés sont particulièrement efficaces lorsqu’ils sont utilisés en association avec d’autres biocides qui attaquent également les parois cellulaires.
La mousse est un problème avec les amines quaternaires. Le chlorure d’alkyldiméthylbenzylammonium (ADBAC) quat mousse beaucoup, mais des composés peu moussants comme le chlorure d’ammonium didécyldiméthyle (DDAC) sont maintenant disponibles. La mousse peut être problématique dans les systèmes d’eau de mer où la mousse peut interférer avec le fonctionnement des tours de dégazage. Les propriétés surfactantes des quats peuvent inhiber la séparation des émulsions huile/eau dans les systèmes de production de champs pétrolifères. L’eau dure peut diminuer l’activité biocide des quats, et les quats peuvent réagir avec des inhibiteurs de tartre et de corrosion chargés négativement, ce qui réduit l’efficacité.
Sulfate de tétrakishydroxyméthylphosphonium (THPS)
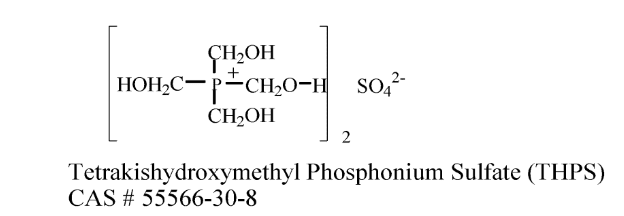
Le THPS est principalement utilisé dans les applications de champ pétrolier, mais il a également été utilisé dans les systèmes de traitement de l’eau traditionnels ainsi que dans certaines applications non biocides. Produit étroitement apparenté, le chlorure de tétrakishydroxyméthylphosphonium (THPC) est un retardateur de flamme courant. THPS est unique en ce sens qu’il tuera les micro-organismes, réduira les concentrations de sulfure d’hydrogène et réagira avec le sulfure de fer et le dissoudra.

TECHNIQUES MODERNES POUR LA PROTECTION CONTRE LA CORROSION/L’ENCRASSEMENT DANS LES TOURS DE REFROIDISSEMENT DE TAILLE MOYENNE
Nettoyage et assainissement des encrassements microbiologiques
L’évolution du remplissage de pellicule de tour de refroidissement à haute efficacité a entraîné une amélioration significative de la performance de la tour. Cependant, comme nous l’avons déjà noté, avec l’augmentation de la surface de remplissage à haute efficacité, le potentiel de formation de tartre et d’encrassement augmente. La meilleure méthode pour nettoyer le remplissage de la tour dépend de plusieurs facteurs, y compris les problèmes de sécurité, la métallurgie du système, le nettoyage en service par rapport au nettoyage hors service, l’impact potentiel sur les opérations de l’usine, les options d’élimination de la solution de nettoyage, l’impact sur l’environnement et la nature chimique et physique de l’encrassement.
Balances minérales
Les dépôts minéraux durs sont le plus souvent composés de silice/silicates ou de carbonate de calcium (calcite). La solubilité de la silice est proportionnelle à la température, et des dépôts se forment souvent près du fond de l’emballage de remplissage à contre-courant où la température de l’eau de refroidissement est la plus basse, les solides dissous sont les plus concentrés et une distribution inégale de l’eau/air peut entraîner des zones sèches. L’eau à température plus élevée en haut du remplissage a la solubilité de calcite la plus faible et favorise une cinétique de dépôt plus rapide. Cependant, à mesure que l’eau passe à travers le remplissage, les minéraux sont légèrement concentrés par évaporation et le pH augmentera légèrement à mesure que l’excès de CO2 est éliminé.
Une technique qui peut être efficace sur le tartre dur aux premiers stades du dépôt est d’appliquer des surfactants spécialisés qui pénètrent les dépôts et induisent l’écaillage du substrat en plastique légèrement flexible. Le surfactant est généralement appliqué avec le programme d’inhibiteur de tartre normal pendant une période prolongée de 60 à 180 jours. Le traitement peut éliminer jusqu’à 70 à 80 % des dépôts. Avant le nettoyage, il est impératif d’identifier et de corriger les conditions de formation de tartre.
Pour les grands systèmes de refroidissement, où le tartre prédominant est calcite, le remplissage peut être nettoyé au fil du temps en réduisant le pH de fonctionnement et/ou les cycles de concentration jusqu’à ce que l’eau soit sous la saturation en calcite. Le calcite sert souvent de liant pour la matrice de dépôt, de sorte que la dissolution du carbonate de calcium peut souvent être très efficace pour décomposer l’ensemble du dépôt. L’acide sulfurique est un choix évident pour les systèmes qui l’ont déjà pour le contrôle du pH. Les composés moins corrosifs comprennent les acides organiques ou l’acide sulfamique inhibé. Certains acides organiques sont plus efficaces que les acides minéraux au pH intermédiaire et sont synergiques avec l’acide sulfurique. À un pH de 5, l’application d’un acide organique approprié accélérera le taux de dissolution de la calcite de 10 à 20 fois par rapport à l’acide sulfurique seul.
Pour le détartrage au carbonate de calcium principalement léger, le nettoyage à la mousse acide hors ligne a été utilisé avec succès, du moins sur les tours de refroidissement plus petites. Lorsqu’il est appliqué par des spécialistes qualifiés en haut du remplissage, le passage lent de la mousse à travers le remplissage prolonge le temps de contact de l’acide avec le tartre. Le volume relativement faible de la solution de nettoyage en mousse utilisée et principalement neutralisée peut potentiellement être recueilli dans le puisard pour l’élimination, ou peut-être mélangé à l’eau circulante des cellules de tour en service voisines. Ce mélange est soumis aux exigences de sécurité et environnementales de l’usine.
Les tartres minéraux peuvent également être enlevés mécaniquement, dans une certaine mesure, in situ ou ex situ. Selon la nature cassante de la balance par rapport au matériau de remplissage flexible en PVC, certains dépôts peuvent généralement être délogés par le nettoyage mécanique du dessous.
Matrices de gisement microbiologiques/organiques
Les dépôts où les microbes ou les matières organiques servent de liant pour la matrice de dépôts sont caractérisés par une consistance douce, souvent semblable à la boue. Contrairement aux tartres minéraux, les dépôts microbiologiques ont tendance à s’accumuler principalement au milieu du remplissage. Les vitesses d’eau directement sous les buses de pulvérisation sont généralement suffisamment élevées pour inhiber l’adhésion microbienne dans la couche supérieure. Pour cette raison, l’encrassement microbiologique passe parfois inaperçu parce qu’il n’est pas visible du haut. À mesure que la vitesse de l’eau ralentit dans le remplissage, les microorganismes commencent à coloniser les surfaces de remplissage, agissant comme filtre pour les solides en suspension. L’encrassement a tendance à être plus intense au milieu qu’au bas, car les derniers pouces de remplissage ne supportent pas physiquement un dépôt épais et mou. L’incapacité de voir clairement le dépôt microbiologique du haut ou du bas, combinée à la difficulté d’inspecter les couches de remplissage du milieu, permet souvent à l’encrassement de progresser sans être détecté jusqu’à ce qu’il atteigne un stade avancé. Plusieurs techniques ont été conçues pour surveiller les zones sensibles, y compris la suspension des sections de remplissage des cellules de charge; la coupe d’une fenêtre d’accès dans le boîtier de la tour pour permettre le retrait périodique d’une section centrale pour l’inspection; la suspension d’une section de remplissage sous le remplissage principal pour permettre son inspection et son pesage faciles. Chaque méthode a été partiellement réussie, mais aucune ne s’est avérée totalement satisfaisante.
Plusieurs méthodes existent pour éliminer les dépôts de noyer biologique du remplissage de la tour de refroidissement. L’hyperhalogénation est une technique courante, mais les résultats sont souvent décevants. La corrosion potentielle des composants du système et la nécessité de déchlorer avant la décharge sont d’autres problèmes importants lorsque vous envisagez cette méthode.
Les dépôts microbiologiques ont généralement une teneur élevée en eau et se rétrécissent et se détachent des surfaces lorsqu’ils sont complètement séchés. Cependant, le séchage efficace du remplissage de la tour de refroidissement peut s’avérer problématique, même avec l’aide de ventilateurs, et même si la tour est située dans un climat à faible humidité. Le dioxyde de chlore a également été utilisé comme nettoyant pour les biofilms des tours de refroidissement avec un certain succès. La méthode de nettoyage la plus répandue et la plus efficace pour les dépôts avec des liants microbiologiques ou organiques est le peroxyde d’hydrogène (H2O2) en raison de sa force d’oxydation et de l’action physique des microbulles d’oxygène produites lorsqu’elles réagissent aux dépôts organiques. Les aspects environnementaux positifs du peroxyde d’hydrogène de la dégradation rapide à l’eau et à l’oxygène, et sa facilité d’application sont des facteurs supplémentaires favorisant le peroxyde comme nettoyant de remplissage. Les doses typiques se situent dans la plage de 500 à 3 000 ppm d’H2O2 actif. L’ajout de surfactant à de faibles concentrations aidera à déloger les dépôts. Les dispersants polymériques sont souvent inclus pour maintenir les solides en suspension pour les rejets d’eaux usées. Les procédures d’élimination doivent être bien établies pour gérer le rejet de ces eaux généralement trouble.
Les figures 7.76 a et b ci-dessous illustrent l’apparence de dépôts de limon sur un remplissage de tour de refroidissement à haute efficacité modérément encrassé avant et après le nettoyage.


Dans les cas où le dépôt contient un pourcentage élevé d’inorganiques, l’eau en circulation peut devenir très trouble. Les procédures du projet doivent inclure des dispositions pour l’élimination des eaux usées trouble après le processus de nettoyage.
Dispersants pour faciliter le nettoyage des dépôts microbiens
Dans les cas où l’encrassement biologique d’une tour de refroidissement ou d’un échangeur de chaleur ne nécessite pas de nettoyage hors ligne, l’alimentation en biodispersant supplémentaire avec le ou les biocide(s) peut être suffisante pour garder l’équipement propre et maintenir des conditions propres.
Un biodispersant efficace avec ses propriétés surfactantes peut aider à prévenir la fixation microbienne aux surfaces, ou peut faciliter l’élimination des dépôts. Plusieurs types de surfactants sont disponibles, y compris les sulfonates anioniques d’alkylbenzène, les polyglycosides d’alkyle et les polymères non ioniques d’oxyde d’éthylène/oxyde de propylène (EO/PO).
Les composés anioniques les plus courants empêchent l’agglomération de particules plus petites en absorbant sur les surfaces de particules, augmentant ainsi la charge de surface négative et la répulsion des particules. Étant donné que ces dispersants ont une charge négative, ils peuvent interagir avec des espèces cationiques dans l’eau (particulièrement le calcium dans les tours de refroidissement hautement recyclées) et perdre de l’efficacité.
Les biodétergents sont généralement des molécules non ioniques qui ne réagissent pas avec d’autres composés de traitement de l’eau. Ils présentent une bonne efficacité dans les eaux dures et sont des composés stables.
Le rôle du dispersant n’est pas de tuer les microbes, mais d’aider à éliminer les dépôts. La concentration initiale du dispersant dépend de l’étendue des colonies existantes et sera plus élevée que les doses d’entretien subséquentes. Le dispersant doit être injecté avant le traitement par biocide, afin qu’il puisse revitaliser les biofilms et augmenter l’efficacité de l’agent(s) tuant. Comme pour tout surfactant, le potentiel de formation de mousse existe, des procédures doivent donc être en place pour combattre les problèmes de formation de mousse. De plus, le choix du dispersant peut être influencé par la toxicité environnementale du produit chimique et la réglementation potentielle dans le permis de décharge.
Dispositifs de traitement non chimiques
Alors que les programmes traditionnels de traitement de l’eau comptent sur diverses chimies pour contrôler le tartre, la corrosion et l’encrassement microbiologique dans les systèmes d’eau industriels, certaines entreprises commercialisent des technologies non chimiques comme alternatives écologiques ou respectueuses de l’environnement. Les dispositifs magnétiques, les systèmes à puissance pulsée, les systèmes électrostatiques, les instruments à ultrasons et les systèmes de cavitation hydrodynamique en sont des exemples. Les fabricants affirment que les dispositifs peuvent remplacer les traitements chimiques traditionnels.
Au début, ces allégations n’ont pas été rigoureusement testées pour déterminer si les appareils étaient effectivement capables des capacités annoncées. Cependant, dans avril 2010, l’American Society of Heating, Refrigerating and Air-Conditioning Engineers (ASHRAE) a publié un rapport de recherche sur plusieurs de ces technologies dans les tours de refroidissement pilotes. Bien que les fournisseurs d’aimants ne fassent souvent pas d’allégations sur le contrôle microbiologique, les autres allèguent souvent une réduction de l’encrassement microbiologique. Le rapport de l’ASHRAE indiquait qu’aucun des dispositifs physiques de traitement de l’eau n’a réduit les populations microbiologiques dans les systèmes de tours de refroidissement pilotes, tandis que les traitements traditionnels à base de chlore étaient efficaces. Une étude subséquente (référence 24) a confirmé que ces mêmes dispositifs non chimiques ne pouvaient pas réduire la légionelle planctonique ou sessile ou les populations microbiologiques globales dans les tours pilotes.
Une méthode non chimique qui a démontré un degré important de contrôle microbien de l’eau de refroidissement est la lumière ultraviolette (UV). Cette technologie continue de s’améliorer, et des travaux récents ont montré que les rayons UV sont particulièrement efficaces (avec une conception de système appropriée) pour le contrôle de l’encrassement macroscopique. Une préoccupation principale avec les rayons UV est la capacité à fournir une énergie suffisante de manière fiable à de grands volumes d’eau ou dans quelle mesure la pénétration des rayons UV est influencée par la clarté de l’eau. Par exemple, la turbidité des solides en suspension peut protéger les micro-organismes de l’énergie UV. Les lignes directrices recommandées pour la sélection des rayons UV dans le cadre d’un programme de contrôle microbiologique sont :
- Turbidité : < 5 NTU
- STS : < 10 mg/L
- Dureté totale généralement égale ou inférieure à une plage de 125 à 200 mg/L, car l’entartrage de la dureté sur les manchons en quartz des lampes peut influencer la transmittance
- Plage de pH : 6,0 à 9,0
La dose d’UV est proportionnelle à l’intensité multipliée par le temps de séjour. La plupart des microorganismes peuvent être détruits avec une dose UV d’environ 10 000 μW-s/cm2. Une recommandation courante est la conception du système avec un dosage UV de 30 000 μW-s/cm2.
Méthodes de contrôle de l’encrassement macroscopique
Les organismes aquatiques tels que les moules zébrées, les palourdes asiatiques et autres développent une coque complète à mesure qu’ils atteignent leur maturité et se fixent aux surfaces où ils filtrent l’eau pour les nutriments. Les organismes adultes peuvent détecter les biocides oxydants et « se résorber » pendant de longues périodes jusqu’à ce que la menace de toxicité soit passée. Si des colonies deviennent établies, des méthodes mécaniques fastidieuses peuvent être nécessaires pour éliminer les organismes qui, lorsqu’ils meurent, produisent des odeurs extrêmement désagréables. Cependant, les organismes peuvent être plus facilement détruits au stade larvaire, avant que les enveloppes protectrices ne se soient complètement développées. Ces jeunes organismes sont connus sous le nom de végétariens. Les sections suivantes examinent les méthodes chimiques et physiques pour contrôler les macro-organismes.
Contrôle chimique : Biocides oxydants
Le chlore est un biocide puissant pour les légumes, et même si de nombreuses installations sont limitées à deux heures d’alimentation en chlore ou en javellisant par jour, si le produit chimique est constamment alimenté chaque jour, de nombreux légumes succomberont. Dans certains cas, les usines ont reçu des dérogations des organismes de réglementation environnementale pour une alimentation continue sur plusieurs semaines afin de tuer les organismes, à une concentration de chlore libre résiduel commune de 0,2 à 0,5 ppm. Pour le contrôle des palourdes asiatiques, une chloration continue pendant quatre semaines au printemps et à l’automne pendant les périodes de pointe peut être efficace. Avec les moules zébrées, la chloration continue pendant l’été peut être efficace. Cependant, les permis d’alimentation continue sont souvent refusés par crainte de formation et de rejet de composés organiques halogénés, appelés trihalométhanes (THM).
Le brome est plus toxique pour les palourdes asiatiques adultes et juvéniles que le chlore, et c’est un oxydant plus rapide qui améliore l’application intermittente. De plus, le brome se dégrade généralement plus rapidement dans l’environnement et est moins corrosif pour le cuivre, les alliages de cuivre et l’acier inoxydable que le chlore.
Le traitement continu au dioxyde de chlore s’est avéré efficace pour le contrôle des moules en zèbre. Le ClO2 nécessite des doses plus faibles et des temps d’exposition plus courts que le chlore. Une concentration de 0,2 ppm offre une mortalité à 100 % des moules en seulement huit jours par rapport à plus de trois semaines ou plus pour le chlore. À 1,0 ppm résiduel, le ClO2 fournira une mortalité de 99 % en une exposition de quatre jours par rapport à 14 jours pour le chlore. À des températures d’eau plus élevées pendant l’été, la toxicité est améliorée et le temps de destruction réduit. Le traitement intermittent avec du dioxyde de chlore nécessite des concentrations résiduelles plus élevées. Un avantage majeur du dioxyde de chlore est la production minimale de THM par rapport au chlore. Les inconvénients comprennent les coûts des produits chimiques et de l’équipement, ainsi que les préoccupations concernant l’entreposage des produits chimiques. La désactivation du dioxyde de chlore résiduel et du chlorite, un sous-produit du procédé, peut être nécessaire avant la décharge pour assurer la sécurité aquatique.
Biocides non oxydants
Plusieurs biocides non oxydants sont approuvés par l’EPA pour les traitements de macrosalissure. Ces biocides sont des composés cationiques qui se sont avérés efficaces pour tuer les palourdes et les moules. Ils comprennent les composés quaternaires décrits précédemment pour le contrôle de l’encrassement microbiologique.
Comparativement aux oxydants, les composés non oxydants ne déclenchent pas de réponse dans les mollusques et les palourdes, et par conséquent, les organismes continuent de filtrer l’eau et sont exposés à des doses chimiques mortelles. Un autre avantage d’un programme non oxydant est que les composés ne nécessitent pas de production sur place et que les systèmes d’alimentation peuvent donc être facilement installés. La chimie ne produit aucun THM, cependant, les composés offrent un risque environnemental et une toxicité potentielle à d’autres organismes aquatiques. Ainsi, ils ne peuvent pas être légalement utilisés sans l’autorisation des organismes de réglementation environnementaux de l’usine, avec les conditions écrites dans le permis de décharge NPDES de l’installation. Le permis nécessitera souvent l’alimentation en argile ou en bentonite du flux de rejet pour adsorber et désactiver les concentrations résiduelles. De plus, comme pour tout produit chimique, les procédures de sécurité lors de la manipulation d’agents non oxydants sont très importantes. Le personnel de l’usine doit porter tout l’équipement de protection du personnel (EPI) requis et doit suivre toutes les procédures de manipulation figurant dans la lettre. Les fiches de données de sécurité (FDS) doivent être disponibles sur le site d’alimentation, avec une deuxième copie située dans un emplacement central comme la salle de contrôle.
Méthodes de contrôle physique
La plupart des admissions d’usine ont des tamis ou des crépines pour empêcher les gros débris de pénétrer dans le système de refroidissement, et ces tamis peuvent efficacement empêcher le passage du mollusque adulte. Cependant, les écrans rugueux ne font que peu pour prévenir l’infiltration du végétarien et la formation subséquente de colonies. Les moules à zèbre présentent une mortalité à des températures supérieures à environ 40 °C (104 °F), qui a été occasionnellement utilisée dans les usines avec une disposition de tuyauterie qui permet la recirculation des décharges d’eau de refroidissement à l’admission. La technique peut être particulièrement efficace en hiver, lorsque les moules sont acclimatées à l’eau froide. D’autres méthodes non chimiques qui ont donné des résultats mixtes comprennent le courant électrique, le rayonnement d’ionisation et la dessiccation (séchage). Les moules zébrées peuvent vivre jusqu’à trois semaines hors de l’eau, ce qui a été un facteur dans leur propagation dans une grande partie du pays. Ils se fixent à une embarcation récréative, et lorsque les bateaux sont transportés d’un plan d’eau à un autre, les moules infestent la nouvelle source.
Aucune méthode mécanique/physique unique ne s’est révélée complètement efficace pour contrôler ou prévenir l’encrassement macroscopique, ce qui souligne l’importance de programmes de traitement chimique bien conçus et exploités pour minimiser ces parasites. Une fois les colonies établies, un arrêt de l’usine peut être nécessaire pour les éliminer mécaniquement.
Méthodes de surveillance microbiologique
Les tests sur le terrain et les analyses en laboratoire central sont utiles pour identifier les microbes et sélectionner les programmes de traitement chimique appropriés. Une identification précise des organismes peut être particulièrement importante pour la sélection des biocides non oxydants. Les méthodes de surveillance des concentrations de biocide dans l’eau de refroidissement sont également importantes pour assurer des résidus appropriés. On ne peut trop insister sur l’importance du contrôle des colonies de sessiles.
Essais sur le terrain du microbes
Diapositives de trempage
Les lames de trempage sont composées d’un support en plastique qui est recouvert de deux côtés avec un milieu de culture de gélose solide qui peut contenir le même média ou un média différent de chaque côté, chacun ayant des indicateurs métaboliques (teintures).
Les supports bilatéraux augmentent la variété de microbes pouvant être identifiés, par exemple les bactéries et les champignons. Une période d’incubation courante pour les lames trempées est de deux jours à la température spécifiée par le fabricant. La population planctonique peut ensuite être estimée en comparant le mat de micro-organismes qui ont grandi sur le nutriment de la gélose à un tableau de référence.
Les avantages des lames trempées sont la facilité d’utilisation et de transport. Une difficulté est que différents fabricants de lames de trempage utilisent différents types de gélose, et certains types peuvent ne pas soutenir la croissance des bactéries spécifiques présentes dans de nombreux systèmes d’eau industriels.
Films Petri
Les pellicules de petri sont des plaques de plastique souples qui contiennent de la gélose, des nutriments et des colorants pour rendre les microbes facilement visibles. En raison de leur taille mince et de leur facilité de manipulation, les films Petri sont adaptables sur le terrain ou en laboratoire. Divers films Petri sont disponibles pour l’identification spécifique des microbes, y compris les bactéries aérobies, E. Coli, la levure, les moisissures et plusieurs autres.

Analyses de l’adénosine triphosphate
L’adénosine triphosphate (ATP) est le principal transporteur d’énergie pour tous les organismes vivants. Parce que l’ATP joue un rôle fondamental et important dans la production d’énergie cellulaire, la régulation métabolique et la signalisation cellulaire; de nombreuses méthodes existent pour mesurer ce composé important. Une méthode courante utilise une enzyme dépendante de l’ATP, la luciférase, pour générer de la lumière. La lumière est captée par un détecteur appelé luminomètre. La réaction presque immédiate permet de calculer presque instantanément la concentration microbiologique. La procédure consiste à mélanger un échantillon d’eau (certaines méthodes reposent sur la concentration des microbes dans un échantillon à l’aide de filtres à membrane) avec un surfactant pour lyser les cellules, libérant ainsi l’ATP dans la solution. La luciférine et l’enzyme de la luciférase sont ensuite ajoutées avec un mélange complet, suivi d’une mesure de la lumière.
Méthodes de laboratoire
Vous trouverez ci-dessous de brèves descriptions de certains des tests microbiologiques de laboratoire les plus courants.
Plaques de gélose spécifiques
Les plaques de gélose sont des boîtes de Petri circulaires contenant un milieu gélatineux enrichi en nutriments pour soutenir la croissance bactérienne. La gélose elle-même peut être fabriquée en laboratoire à partir de recettes et appliquée sur les assiettes. Des plaques de gélose pré-coulées sont également disponibles. Les plaques de gélose peuvent être non sélectives pour la croissance générale ou peuvent contenir des ingrédients spécifiques qui favorisent la croissance d’un organisme cible comme Pseudomonas ou Enterobacteriaceae.
Tests BART
Les tests de réaction d’activité biologique (BART) sont une méthode simple, sur le terrain ou en laboratoire, de détection de certaines bactéries dans les échantillons d’eau. Les échantillons sont incubés sur plusieurs jours. Chaque BART possède un indicateur visuel unique qui permet à l’utilisateur d’identifier la présence ou l’absence de bactéries spécifiques. La population cible de microbes est estimée à partir du nombre de jours d’incubation jusqu’à ce que la couleur ou l’opacité apparaisse dans le tube.

Les BART sont efficaces pour identifier les bactéries liées au fer, réduisant le sulfate, formant la slime, nitrifiantes et dénitrifiantes. La méthode est essentiellement un test de type présence/absence et ne fournit pas de données démographiques. Les tests peuvent être pratiques dans les applications de champ pétrolifère pour la détection générale des bactéries anaérobies sans avoir besoin d’équipement coûteux.
Cytométrie en flux
La cytométrie en flux est une technique d’analyse des particules qui utilise la lumière laser pour analyser les caractéristiques physiques et chimiques des particules dans un flux de fluide. La taille et la quantité des particules en suspension sont mesurées en analysant l’effet d’éclipse que les particules produisent lorsqu’elles passent devant le laser. La quantité de lumière diffusée est associée à la complexité des particules. Les particules peuvent être comptées et triées en fonction de leur taille et de leur complexité. Les avantages de la cytométrie en flux comprennent la spécificité de l’analyse, l’analyse simultanée d’un échantillon pour plusieurs types de bactéries et la capacité d’obtenir des résultats en quelques heures plutôt qu’en quelques jours. Cette technologie est principalement employée dans l’industrie médicale.
Microscopie fluorescente – Coloration sous tension/morte
La microscopie est une méthode bien établie pour analyser visuellement les bactéries. La coloration fluorescente peut différencier diverses bactéries et, avec l’utilisation de combinaisons de colorants, peut identifier les bactéries vivantes et mortes. L’application d’une tache vivante ou morte sur un biofilm peut aider à confirmer si le programme de traitement chimique est efficace pour pénétrer le biofilm et tuer les bactéries à l’intérieur. Bien que la coloration sous tension/à vide soit une excellente technique visuelle et offre une méthode rapide d’analyse d’un échantillon, elle nécessite un microbiologiste formé, un environnement contrôlé et des instruments et réactifs spécialisés.
Analyses des microorganismes Sessile
La plupart des techniques énumérées ci-dessus concernent la spéciation des organismes planctoniques. Si des colonies de sessile se développent et sont accessibles pendant les inspections hors ligne, les inspecteurs doivent recueillir des échantillons pour des analyses de laboratoire. Ils peuvent fournir des informations précises sur les organismes qui prospèrent dans cet environnement, ce qui offre à son tour des conseils pour les modifications du programme de traitement chimique.
*N.B. :Pour tout personnel qui pourrait entrer en contact avec des colonies de sessiles ou respirer l'atmosphère autour d'eux lors des inspections ou des nettoyages d'échangeurs de chaleur, il existe un risque d'exposition à la bactérie legionella ou à d'autres microorganismes nocifs. Un équipement de protection individuelle (EPI) approprié, y compris des masques, est indispensable.
Surveillance des biocides oxydants
Une analyse régulière du biocide résiduel oxydant est extrêmement importante pour assurer des doses constantes et appropriées.
La méthode DPD
Avant le développement de méthodes de surveillance en ligne, la procédure standard pour la mesure de l’halogène libre ou total était la méthode DPD. DPD est l’acronyme de N,N-diéthyl-p-phénylènediamine. Il réagit avec les oxydants dans l’eau pour former un complexe coloré, dont l’intensité est directement proportionnelle à la quantité d’oxydant présente.
Il est à noter que la DPD réagit avec les oxydants standard, y compris les résidus libres et combinés. La monochloramine est un bon exemple de cette dernière. Les analyses libres ou combinées nécessitent un facteur de multiplication différent pour convertir la couleur observée en concentration. Une autre variable est le temps de réaction. Les résultats des tests de résidus libres sont généralement disponibles en moins de 30 secondes, tandis que le test du résidu total nécessite généralement 3 à 6 minutes pour une réaction complète. Chacun nécessite une trousse de test distincte. Bien que les deux comprennent la DPD et un tampon de pH, la trousse résiduelle totale comprend également de l’iodure de potassium et un tampon supplémentaire. Ces deux composés améliorent la formation de la couleur et permettent aux oxydants moins réactifs de se combiner au DPD.
Analyse du chlore en ligne
Une mesure en ligne du biocide oxydant mature est disponible. Un instrument industriel standard est illustré ci-dessous.

L'analyseur extrait un échantillon, et selon la discussion DPD ci-dessus, injecte le colorant et le tampon. Après le temps de réaction approprié, l'instrument affiche la concentration de l'oxydant, purge l'échantillon dépensé et extrait un nouvel échantillon. Ainsi, l'analyseur peut fournir des analyses 24 heures sur 24, séparées uniquement par le temps nécessaire à la réaction du réactif. La surveillance en ligne peut être très utile pour détecter rapidement les défaillances du système d’alimentation en biocide ou les changements de la chimie de l’eau qui augmentent la demande en biocide. Tout comme les analyses DPD avec échantillon, l'analyseur dispose d'un réactif résiduel libre ou total.
Potentiel d’oxydation et de réduction
Les eaux de refroidissement contiennent généralement un certain nombre de composés oxydants ou réducteurs. La mesure du potentiel de réduction de l’oxydation (PRO) est une technique analytique qui, comme son nom l’indique, fournit des données concernant l’oxydation ou la réduction de la puissance de la solution. Lorsque des biocides oxydants sont utilisés pour le contrôle microbiologique, leur potentiel oxydant éclipse généralement les autres espèces.
Les sondes ORP courantes sont généralement dotées d’une électrode fonctionnelle contenant un fil en platine et d’une électrode de compteur en argent/chlorure d’argent scellée. Ces sondes sont plutôt robustes et peuvent souvent survivre pendant plusieurs années dans des conditions normales de fonctionnement de l’eau de refroidissement et de l’eau de traitement.
Certaines mises en garde existent avec ORP pour la surveillance des résidus de biocide. Les mesures ne sont pas linéaires, surtout à de faibles concentrations. La température influence le rH; et la compensation de température, qui nécessite une sonde supplémentaire, est importante pour des lectures précises.
Même avec ces mises en garde, les mesures du TRO peuvent tout de même fournir des informations significatives concernant les concentrations de biocides. En présence d’un biocide oxydant, le PRO sera généralement élevé par rapport aux valeurs de référence à une plage de 300 à 700 mV, selon les espèces d’oxydants, la concentration, la température, le pH et la présence d’autres espèces. Le rH peut vérifier que l’oxydant est présent et détectera également si les conditions passent d’aérobie (rH positif) à anaérobie (rH négatif). De tels changements peuvent indiquer un afflux de contaminants grave.
Surveillance de l’alimentation, des dépôts et de la corrosion des produits chimiques
Les sections précédentes décrivent les mécanismes de dépôt et de corrosion de l’eau de refroidissement et les méthodes de contrôle. Les sections suivantes décrivent les techniques de contrôle des dépôts et de la corrosion.
Surveillance des dépôts
Le chapitre 6 offrait une méthode de surveillance indirecte; c’est-à-dire l’évaluation de la performance de l’échangeur thermique par l’entremise des températures d’entrée, de sortie et de fluide de procédé; et à partir de ces lectures et d’autres, les calculs en ligne du transfert de chaleur. Grâce à des algorithmes modernes, les données peuvent être continuellement orientées pour permettre une détection rapide de la formation de tartre ou de l’encrassement microbiologique.
Les coupons de dépôt offrent une autre option.

Ces coupons peuvent être inclus avec les coupons de corrosion dans un système d’échantillonnage de dérivation pour recueillir les matières en suspension et les tartres minéraux. L’extraction périodique et les examens visuels fournissent des données sur la formation de tartre (ou son absence). Les mesures de densité de poids et les analyses de laboratoire de la composition du dépôt peuvent fournir des données précieuses.
Les photographies des coupons immédiatement après l’extraction sont importantes pour documenter les conditions des coupons. Un problème avec les coupons est qu’ils n’atteignent pas la température de surface des tubes de l’échangeur de chaleur, et par conséquent, le dépôt ou la corrosion peut être plus doux qu’il n’existe réellement dans certains endroits du système. Deux outils qui simulent plus précisément les conditions réelles sont le moniteur de dépôt et le test de l’échangeur de chaleur.

Ces unités contiennent des tubes d’échangeur de chaleur amovibles. L'eau de retour (chaude) du système de refroidissement est l'alimentation, avec une tige chauffée dans le compartiment à échantillons. Cette configuration place théoriquement l’échantillon du tube sous la contrainte la plus élevée de tout échangeur de chaleur dans l’ensemble du système de refroidissement. Si des conditions de détartrage sont présentes, un dépôt doit d’abord se produire dans l’unité d’essai. Inversement, si le programme de traitement chimique empêche la formation de tartre dans l’unité d’essai, on peut raisonnablement supposer que le reste du système est propre. Un avantage évident est que si un détartrage se produit, le tube peut être facilement retiré pour l’analyse des dépôts. L’une des clés de la réussite de l’application de cette technologie est une étude complète du système de refroidissement pour s’assurer que les conditions de l’unité d’essai sont plus sévères que celles de tout échangeur de chaleur de procédé.
Surveillance de la corrosion
La méthode de surveillance de la corrosion la plus fondamentale est par les coupons de corrosion. Les coupons doivent être faits des mêmes métaux que ceux qui se trouvent dans le système de refroidissement. Les coupons simuleront les mêmes taux de corrosion que les composants du système, mais avec la mise en garde de nouveau, ils ne représentent pas entièrement les conditions sur les surfaces des tubes ou des plaques, en particulier en ce qui concerne les influences de température.
Le coupon le plus fréquemment utilisé pour la surveillance de la corrosion de l’eau de refroidissement est le C1010 (acier doux), mais les autres matériaux courants comprennent l’acier inoxydable (généralement un ou plusieurs de la série 300), le laiton admiral, le nickel-cuivre, l’aluminium, l’acier galvanisé et toute autre métallurgie qui pourrait être présente.
Les coupons de corrosion neufs, nettoyés et prépesés doivent être mis en service pendant au moins 30 jours, avec des périodes plus longues de 60, 90 et 120 jours également recommandées. Cette plage fournit des données sur la corrosion à court et à long terme.
L’apparence du coupon lors de l’extraction initiale montre le type et la quantité d’encrassement de surface. Cet encrassement peut révéler diverses quantités de produits de corrosion, ou peut-être d’autres débris. Les dossiers et les photographies lors du retrait sont importants, car les dépôts ou les matériaux de corrosion peuvent se déloger pendant l’expédition au laboratoire. Même les odeurs, comme le sulfure d’hydrogène, peuvent être utiles pour identifier un mécanisme d’encrassement. L’inspection visuelle initiale suggère souvent un certain mécanisme de corrosion, mais le nettoyage des coupons en laboratoire et des analyses supplémentaires peuvent fournir des détails supplémentaires concernant les mécanismes de corrosion. Idéalement, bien sûr, les coupons resteront dans un état presque immaculé, signifiant la sélection et la performance appropriées des programmes de traitement chimique.
La figure ci-dessous présente un rapport de corrosion fondamental.
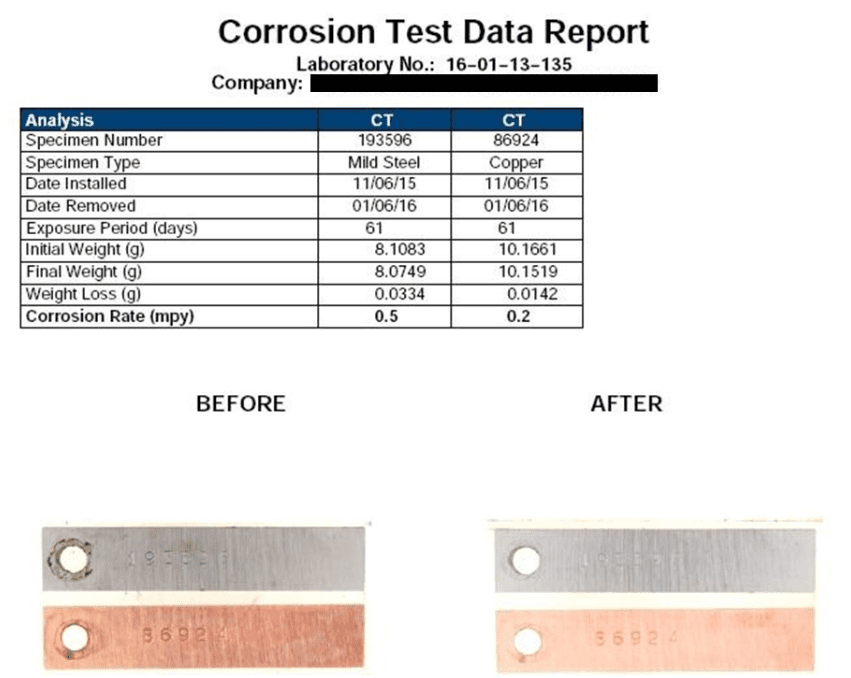
Le rapport et les photos illustrent un coupon de corrosion de l’acier doux et du cuivre soumis à l’environnement de refroidissement de l’eau pendant un peu plus de deux mois. Seule une très légère corrosion générale a été détectée, avec de faibles taux de corrosion de 0,5 et 0,2 mpy, respectivement. Les résultats du coupon suggèrent un excellent contrôle de la corrosion.
Conception de l'échantillonnage des coupons de corrosion
La figure suivante illustre une disposition commune d’échantillonnage de corrosion, le « rack de dérivation de coupon de corrosion » ( rack de coupons). Le matériau de tuyau le plus courant pour l’échantillonnage de l’eau de refroidissement est le PVC-C, mais un tuyau en acier peut être nécessaire pour les systèmes d’eau chaude ou sous pression.

Une des principales caractéristiques de la bonne conception est l’orientation du coupon. Comme il est évident ci-dessus, l’orientation est avec le débit d’eau le long et non contre le coupon. Cette configuration aide à minimiser les courants d’alerte. Le porte-coupon a généralement une « marque témoin » sur la tête hexagonale pour assurer une bonne orientation.
Le porte-coupon est électriquement inerte pour prévenir tout effet galvanique. Dans les systèmes chauds avec tuyauterie métallique, la vis et le boulon doivent être de la même métallurgie que le coupon, avec des connexions séparées par des rondelles non métalliques.
La configuration de la tuyauterie de la Figure 7.85 montre quatre positions potentielles de coupon. Cet arrangement permet un échantillonnage séparé de 30, 60, 90 et 120 jours, ou toute autre fréquence souhaitée, et avec différents métaux. Il est important que différents métaux soient disposés selon la série galvanique du moins au plus noble. L’installation de vannes qui permettent un bon contrôle du débit d’échantillon est également importante avec ces systèmes. Il est recommandé d’utiliser des robinets-vannes plutôt que des robinets-vannes.
Un débat sur l’utilisation de bons-rabais prétraités ou non est souvent soulevé. L’un ou l’autre peut produire des données bénéfiques. Si la préoccupation est de déterminer l’efficacité avec laquelle les concentrations normales d’inhibiteurs dans l’eau en vrac peuvent passiver le système, alors les coupons non traités sont meilleurs. Cependant, si la préoccupation concerne le potentiel de corrosion à court terme, comme les piqûres, alors les coupons prétraités peuvent être meilleurs pour que la corrosion initiale ne masque pas d’autres problèmes.
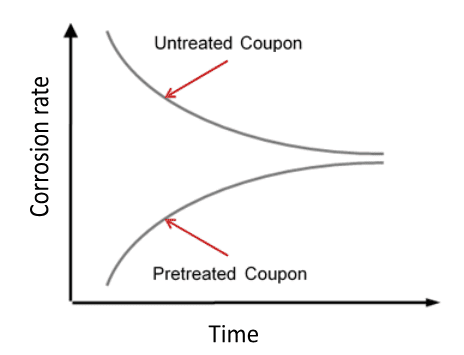
Le coupon non traité subira une corrosion rapide initiale, après quoi le taux diminue à mesure que le coupon devient passivé. Le coupon prétraité a de faibles taux de corrosion initiale jusqu’à ce que la pellicule protectrice de l’inhibiteur atteigne l’équilibre avec le système en vrac, auquel moment le coupon imitera les conditions du système d’eau. Sur une période prolongée (60 à 90 jours), les taux de corrosion s’approchent.
Débitmètres de corrosion en ligne
Des compteurs de corrosion sont disponibles pour mesurer électroniquement la corrosion. Les avantages sont des mesures instantanées et continues. Cependant, ils sont beaucoup plus chers que les coupons de corrosion.
Deux principaux types de compteurs de corrosion sont courants : la résistance de polarisation linéaire (LPR) et la résistance électrique (ER). Cette section se concentre sur la LPR, qui mesure les lectures en milli-tension réelles produites pendant la corrosion.
Résistance de polarisation linéaire
L’instrument peut être équipé d’électrodes du métal d’intérêt, qui se corrodent ensuite par les mécanismes normaux.

La technologie s’applique à l’eau de refroidissement et à d’autres systèmes d’eau, notamment :
- Récupération secondaire de l’huile
- Systèmes de traitement et de distribution de l’eau potable
- Traitement des eaux usées
- Fabrication de pâtes et papiers
- Production d’hydrocarbures avec eau libre
Comme l’indiquent les figures, les sondes à 2 et 3 électrodes sont courantes. Les compteurs LPR peuvent instantanément mesurer les taux de corrosion de 0,01 à 200 mils par an (MPY) et les piqûres de 1,1 à 100 unités de piqûre en utilisant un facteur de proportionnalité qui relie les caractéristiques de polarisation électrique de la sonde à son taux de corrosion.
Tests de corrosion en laboratoire
Les techniques électrochimiques, comme la polarisation cyclique, sont courantes en laboratoire pour évaluer la sensibilité des matériaux à la corrosion localisée. Les paramètres critiques tels que le potentiel de corrosion, le potentiel de piqûre, le courant de corrosion et les potentiels de repassivation peuvent être déterminés à partir de l’expérience de polarisation cyclique.
Le schéma de la configuration électrochimique et les graphiques représentatifs de polarisation cyclique sont illustrés ci-dessous.
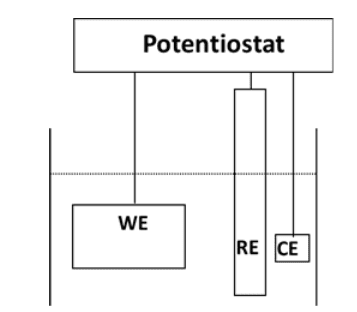
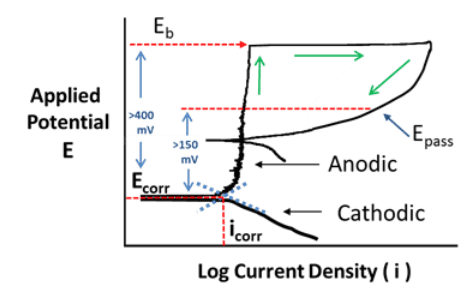
À partir des graphiques de polarisation cyclique, des paramètres importants peuvent être calculés tels que :
- Potentiel de piqûre (Epit) = Eb – Ecorr | Éq. 7-32
- Potentiel de repassivation (ERP) = Epass – Ecorr | Éq. 7-33
Généralement accepté, une valeur Epit supérieure à 350-400 mV associée à un PIU supérieur à 150 mV signifie qu’il existe une possibilité minimale de corrosion localisée et que l’alliage convient à une utilisation à long terme dans cet environnement.
La figure 7.92 illustre les courbes de polarisation cyclique d’un échantillon d’acier inoxydable 304 à Richmond, en Virginie, à une température de 150 °F et avec une concentration de chlorure augmentée à 750 ppm. Divers paramètres critiques de corrosion peuvent être dérivés de ce graphique et sont présentés dans le tableau 7-4. Les données décrivent la protection contre la corrosion fournie par la chimie RPSI de ChemTreat.
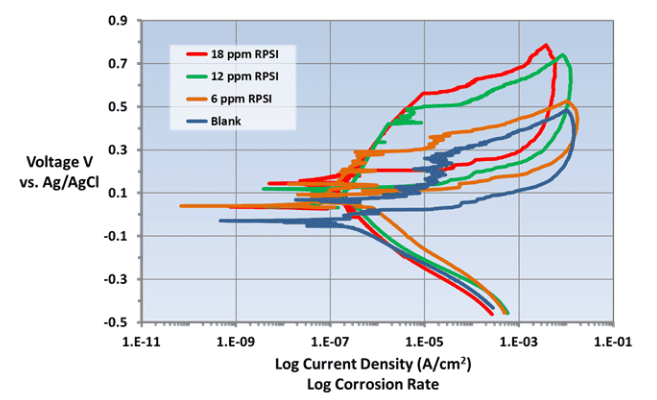
Tableau 7-4.
Divers paramètres de corrosion calculés à partir des courbes de polarisation cycliques illustrées à la Figure 7.92.

Il est clair à partir de ces données que les potentiels de piqûre et de repassivation augmentent avec l’augmentation du dosage du RPSI. 100 ppm RPSI satisfait aux exigences d’Epit supérieures à 400 mV et ERP supérieures à 150 mV, et confirme que le Type 304 SS ne subit pas de corrosion localisée dans ces conditions.
On sait également que certains aciers, et en particulier les aciers inoxydables 304 et 316, peuvent souffrir de fissures de corrosion sous contrainte (SCC) avec des concentrations de chlorure normalement faibles, mais avec une température croissante. La capacité du RPSI à potentiellement protéger le 304 SS contre le CS a été démontrée lors d’essais en laboratoire, au cours desquels des échantillons soumis à une contrainte de courbure en U ont été placés pendant 15 jours dans un bain d’eau du robinet à 105 °C avec un pic de 1 000 mg/L de chlorure. Les figures ci-dessous montrent l’état des échantillons après le test.

Le spécimen non traité est clairement terni et piqué. L’échantillon exposé à la même solution contenant l’inhibiteur de corrosion RPSI ne montre aucun signe de ternissement ou de piqûre. Sous grossissement, la fissuration est clairement évidente à la courbure de l'échantillon non traité alors que l'échantillon traité ne montre aucune fissuration.
Nettoyage et passivation du système de refroidissement
Cette section présente les directives générales de passivation pour les systèmes de refroidissement. Trop souvent, les ingénieurs de conception et le personnel de l’usine considèrent les nettoyages préopérationnels et la passivation du système comme des articles mineurs ou inutiles. Dans ces cas, la procédure peut tout au plus consister à remplir le système d’eau, à effectuer des tests hydrostatiques (le cas échéant), puis à démarrer l’unité. Souvent, le fournisseur de traitement de l’eau est amené dans la discussion beaucoup trop tard pour offrir des informations et des conseils sur la criticité du prénettoyage et de la passivation appropriés. Cette discussion décrit les recommandations générales pour les applications courantes, en sachant que chaque système de refroidissement a des caractéristiques et des métallurgies individuelles. Il est nécessaire de consulter des experts réputés en traitement chimique pour élaborer des procédures de nettoyage et de passivation précises.
Nettoyage pré-opérationnel et passivation
Les nouveaux systèmes de refroidissement contiennent souvent des débris laissés par les phases de construction et d’installation, ainsi que des résidus d’huile et de graisse et des produits de corrosion dus à la rouille instantanée. Il est primordial d’avoir un plan de nettoyage et de passivation en place et effectué avant la mise en service réelle du système. La discussion suivante est divisée en trois catégories.
- Systèmes de recirculation ouverts (pas de composants en acier galvanisé)
- Systèmes de recirculation ouverts (acier galvanisé présent)
- Systèmes de refroidissement fermés axés sur les refroidisseurs
Un aperçu beaucoup plus détaillé des procédures de nettoyage, de passivation et de mise en place est disponible dans la référence 28.
Nettoyage pré-opérationnel et Passivation - Directives générales
En plus du limon, de la saleté, des huiles et de la rouille, de nouvelles tours de refroidissement peuvent également accumuler de la boue organique et des algues dans les puisards et les terrasses qui ont été inondés pour la protection contre les incendies. Ces organismes peuvent encrasser et boucher les tubes du condenseur et de l’échangeur de chaleur, le remplissage de la tour de refroidissement et d’autres équipements.
Directives générales pour tous les nettoyages/passivations
Avant de commencer la procédure de nettoyage et de passivation :
- Rincez et rincez toutes les conduites, puisards et terrasses de tour avec de l’eau propre.
- Assurez-vous que toutes les crépines, tous les filtres, toutes les soupapes inverseurs de débit et tous les drains sont installés et en parfait état de fonctionnement.
- Les pompes d’alimentation en produits chimiques doivent être installées et opérationnelles; cependant, les produits chimiques de nettoyage et de passivation peuvent être alimentés manuellement, au besoin.
- Débranchez ou contournez toutes les sondes de conductivité, de pH et autres sondes analytiques pour éviter l’encrassement et la contamination de cet équipement.
- Préparer une liste de contrôle complète, puis étiqueter toutes les vannes de régulation de débit, les drains et les contrôleurs. S’assurer que tous les équipements ont les bons réglages de vanne (habituellement complètement ouvert ou complètement fermé) avant le nettoyage. Les procédures écrites doivent fournir un guide pour les positions de vanne et autres réglages à chaque étape du processus.
- Les opérateurs d’usine et/ou le personnel d’entretien doivent faire partie d’une équipe dédiée au fonctionnement des vannes, au nettoyage des crépines et des filtres, etc., tout au long du processus. Une réunion de sécurité et de planification/examen est nécessaire au début de chaque quart de travail pendant le processus.
- Les tours de refroidissement et autres équipements ne doivent pas être soumis à des charges thermiques pendant le processus.
Remarque :Des dispositions doivent être mises en place avant le nettoyage pour que les drains et les égouts, les étangs de retenue ou toute autre élimination des déchets de nettoyage soient éliminés. Bien que ces eaux usées ne soient généralement pas dangereuses, elles contiennent généralement des niveaux inacceptables de solides et d’huiles en suspension. De plus, le pH de la décharge peut dépasser les limites de la NPDES de l’usine ou de la POTW (travail de traitement de propriété publique). Des pénalités sévères peuvent être imposées en cas de rejet non autorisé et inapproprié des eaux usées. Souvent, la solution de nettoyage utilisée peut être envoyée à un POTW si des dispositions ont été prises au préalable.
Procédures de nettoyage pour les tours non galvanisées
Les étapes de nettoyage et de passivation peuvent être effectuées ensemble ou en succession immédiate. Dans la plupart des cas, le système d’eau de la tour est d’abord nettoyé et rincé, puis passivé avec de fortes concentrations d’inhibiteurs avant de mettre le système en fonctionnement. Les étapes suivantes sont courantes :
- Remplissez le puisard et les conduites de la tour à des niveaux de fonctionnement normaux et commencez à faire circuler l’eau de la tour à des taux de pompage normaux (en fonctionnement).
- Les échangeurs de procédé, les refroidisseurs et/ou les condenseurs doivent être contournés du côté du procédé. Toutes les entrées et sorties du bord de l’eau doivent être complètement ouvertes.
- Ajouter des produits chimiques inhibiteurs après le début de la circulation. Les produits chimiques peuvent être pompés, ou ébréchés manuellement, dans le puisard de la tour ou injectés directement dans tout collecteur d’alimentation ou de retour d’eau majeur.
En général, les produits chimiques utilisés pour le nettoyage initial et la passivation comprennent des agents pour maintenir un pH neutre ou légèrement alcalin, des surfactants pour l’élimination de l’huile et de la graisse et un agent antimousse. Un pyrophosphate pour maintenir un pH de 10 peut être bénéfique pour améliorer la passivation.
Une fois les produits chimiques ajoutés, l’eau de la tour doit circuler à des taux de fonctionnement normaux pendant quatre à huit heures pour assurer une distribution et un nettoyage appropriés. Les procédures correctement appliquées produiront de l’acier passivé.
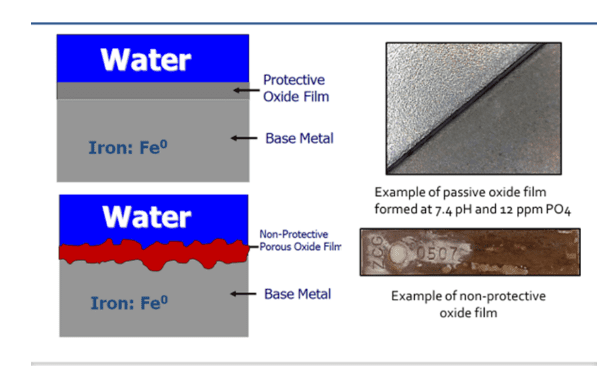
À la fin du processus, la tour, la tuyauterie du système de refroidissement et les échangeurs de chaleur doivent être vidangés et rincés aussi soigneusement que possible pour éliminer les huiles, les limons et les boues biologiques du système. Encore une fois, des dispositions devraient déjà être en place pour éliminer ces eaux usées.
Si le système de refroidissement reste rempli, mais en attente pendant plus de trois jours, une protection avec un biocide non oxydant est recommandée. Common est un produit à base d’isothiazoline sans cuivre. Au minimum, l’eau de la tour doit circuler pendant une à deux heures chaque semaine avant le démarrage, mais une à deux heures par jour est idéale.
Recommandations de prétraitement, de passivation et d’entretien pour les tours de refroidissement galvanisées et les condenseurs à évaporation
Pour les systèmes contenant des composants galvanisés, des procédures spéciales sont nécessaires pour s’assurer que les matériaux galvanisés développent une couche de passivation appropriée. Le tableau ci-dessous présente les recommandations de trois fabricants de tours de refroidissement.
Tableau 7-5 :
Valeurs et plages recommandées pour la chimie de l’eau des fabricants d’équipement d’origine de la tour de refroidissement galvanisée. Source : « White Rust Prevention – An Industry Update and Guide Paper – 2012 », Association of Water Technologies (AWT).
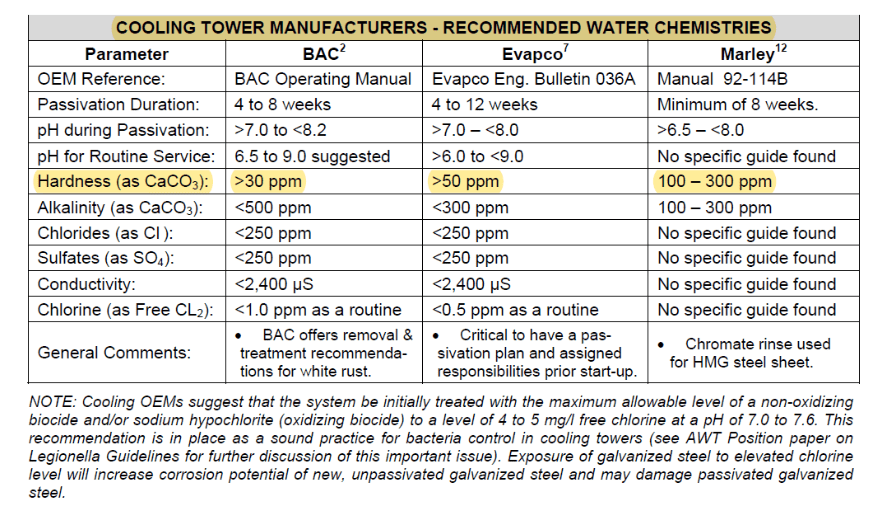
Notez qu’il existe une certaine variabilité dans les lignes directrices de chimie recommandées, mais chaque programme est conçu pour produire un composé de carbonate de zinc hydraté serré sur la surface métallique. On croit que la formule chimique de ce composé est 3Zn(OH)2∙ZnCO3∙H2O.
Si ce conditionnement est omis, ou si une opération subséquente élève l’eau de refroidissement à un pH élevé, le zinc réagira avec l’eau pour former un hydroxyde de zinc gélatineux poreux, qui réagit ensuite avec le carbonate et convertit le matériau en un dépôt blanc et poreux connu sous le nom de « rouille blanche ».

Si de la rouille blanche se développe sur du métal galvanisé, les dépôts deviennent de plus en plus poreux avec une perte croissante de métal en dessous. Cette corrosion exposera éventuellement l’acier au carbone sous-jacent à une attaque.
Une fois que le revêtement passif a été correctement établi, plusieurs fabricants de tours de refroidissement recommandent une plage de pH de fonctionnement normal de 6,5 à 9,0. Cela est bien conforme aux lignes directrices communes de contrôle des dépôts et de la corrosion discutées ci-dessus. Les ressources pour obtenir de plus amples renseignements comprennent le Cooling Technology Institute et des fabricants de tours de refroidissement spécifiques.
Nettoyage pré-opérationnel d’une boucle fermée de refroidisseur
À la suite d'un arrêt prolongé ou d'une nouvelle installation, la rouille instantanée et les conditions stagnantes peuvent avoir un impact considérable sur l'état des réseaux de refroidisseurs. Le fait de ne pas nettoyer et passiver correctement un système avant le démarrage peut entraîner des défaillances répétées imprévues. S’il est possible de prendre des photos des principales conditions de l’équipement avant et après le nettoyage, les photos permettront d’évaluer l’efficacité du nettoyage. Une connaissance complète de la métallurgie du système est essentielle pour sélectionner les produits chimiques de nettoyage et de passivation appropriés. L’acier au carbone constitue généralement la métallurgie du système primaire, mais des alliages de cuivre et de l’acier inoxydable peuvent également être présents. Si des composants en aluminium sont présents dans le système, des précautions particulières sont nécessaires pour protéger ce métal.
Procédure générale de pré-nettoyage
- Établir la circulation du système et rincer l’équipement pour éliminer les gros débris au débit maximal possible. Remplissez le système jusqu’à un volume de conception maximal de 80 % pour permettre l’expansion thermique et l’ajout d’agent de nettoyage. Ne pas prévoir d’expansion peut entraîner des défaillances de l’équipement.
- Injecter les produits de nettoyage/passivation. Ceux-ci consistent en des détergents alcalins, des surfactants et des tampons de pH pour éliminer l’huile et la graisse et d’autres débris, et pour passiver les métaux. Un dérivé azolé peut être inclus pour la passivation de l’alliage de cuivre. Faites circuler la solution de nettoyage dans les échangeurs pendant 12 à 36 heures pour éliminer les produits de corrosion, l’huile et la graisse, et pour passer tous les métaux. Ayez des produits chimiques antimousse à portée de main.
- Lorsque la période de circulation est terminée, vidanger le système pour éliminer les produits chimiques de prénettoyage, les produits de corrosion, le tartre, la saleté, etc. Rincez toutes les vannes et inspectez les pattes mortes. (Idéalement, les pattes mortes doivent être identifiées et éliminées dans la mesure du possible avant le nettoyage, la passivation et le fonctionnement normal.)
- Ajoutez des inhibiteurs de corrosion appropriés et possiblement un biocide non oxydant si l’équipement est en attente après le processus de nettoyage/passivation.
Le processus de nettoyage/passivation est différent pour chaque application et peut être très spécifique pour certaines installations comme les centres de traitement des données.
Mise en place du système de refroidissement
Dans de nombreuses installations, l’équipement fonctionne 24 heures sur 24, 7 jours sur 7, avec peu de temps d’arrêt. Cependant, dans les cas où les systèmes doivent être retirés de la ligne, des procédures de mise en place bien conçues et menées sont nécessaires pour prévenir la corrosion grave. La combinaison d’eau froide et d’oxygène peut être très destructrice pour certains alliages, notamment l’acier au carbone. De plus, les micro-organismes peuvent se déposer et proliférer dans l’eau stagnante. Les défaillances subséquentes des tubes ou des tuyaux de la CMI ont été bien documentées.
Si possible, le drainage et le séchage de l’équipement sont souvent la meilleure méthode pour minimiser la corrosion. La circulation d’air déshumidifiée (DHA) peut être très efficace pour le séchage. Un exemple de ce processus est décrit au chapitre 4 pour la protection des turbines à vapeur pendant les pannes. Cependant, souvent, l’eau doit rester dans les systèmes pour pouvoir être commencée à court préavis. Dans ces cas, plusieurs étapes sont recommandées pour la protection du métal. Ceux-ci comprennent :
- Avoir des procédures de mise en place bien élaborées à portée de main et facilement accessibles au personnel de l’usine. Les détails des procédures doivent comprendre :
- Concentrations d’agents passivants et de biocides à établir dans le réseau.
- Points d’échantillonnage et fréquence des tests pour ces composés
- Fréquence, durée et débits de circulation d’eau (la circulation est très importante pour prévenir la formation d’eau stagnante.)
- Étapes pour rincer les produits chimiques du système, si nécessaire, avant le démarrage de l’unité.
- Méthodes d’élimination des eaux usées
- Directives pour l’inspection de l’équipement. Ceux-ci doivent inclure des procédures d’entrée dans un espace clos.
Conclusion
La chimie de l’eau de refroidissement et la minimisation de la corrosion, de l’entartrage et de l’encrassement microbiologique sont des sujets complexes. De nombreux facteurs influencent ces phénomènes. ChemTreat espère que ce chapitre fournira des informations utiles aux propriétaires d’usines, aux opérateurs et au personnel technique dans le développement de programmes efficaces de traitement de l’eau de refroidissement.
Trouver des solutions de traitement de l’eau personnalisées avec ChemTreat
ChemTreats s’engage à fournir des solutions de traitement de l’eau expertes. Nos solutions de traitement de l’eau innovantes et personnalisées aident à contrôler la corrosion, le tartre et l’encrassement biologique dans les systèmes de refroidissement dans une variété d’industries. Communiquez avec ChemTreatpour communiquer avec votre représentant local afin d’apprendre comment ChemTreat peut aider votre entreprise dès aujourd’hui.
Références
1. W.D. Callister, Jr., Science et ingénierie des matériaux : une introduction, 3e éd., John Wiley & Sons, 1994.
2. Correspondance par courriel avec Dan Janikowski de Plymouth Tube Company, janvier 2022.
3. Post, R. et Kalakodimi, P., « The Development and Application of Non-Phosphorus Corrosion Inhibitors for Cooling Water Systems »; présenté au World Energy Congress, Atlanta, Géorgie, octobre 2017.
4. Notions de base sur la corrosion : Introduction. National Association of Corrosion Engineers, Houston, TX, 1984.
5. Deacon, J. (sans date) The Microbial World : Thermophilic microorganisms, University of Edinburgh, School of Biological Sciences. Université d’Édimbourg, School of Biological Sciences. Disponible à : http://archive.bio.ed.ac.uk/jdeacon/microbes/thermo.htm (Accès : Le 22 novembre 2022).
6. J.W. McCoy, microbiologie de l’eau de refroidissement. Chemical Publishing Company, NY 1980, p. 197–198.
7. Quelle est la différence entre la moisissure et la levure? – quora (sans date) Quora. Disponible à : https://www.quora.com/What-is-the-difference-between-mold-yeast (Accès : Le 1 septembre 2022).
8. Stewart, P.S., 1996. « Aspects théoriques de la diffusion des antibiotiques dans le biofilm microbien », Agents antimicrobiens et chimiothérapie, novembre 1996, p. 2517–2522.
9. Département de la Santé et des Services sociaux des États-Unis. (s.d.). Maladie des légionnaires – à propos de la maladie. Centre d’information sur les maladies génétiques et rares. Page consultée en 2021, à l’adresse https://rarediseases.info.nih.gov/diseases/6876/legionnaires-disease/
10. T. Rowbotham « Rapport préliminaire sur la pathogénicité de la Legionella pneumophila pour l’eau douce et l’amoebae du sol » J. Clin. Pathol. 1980, 33, 1179-1183.
11. J. B. Kurtz, C. L. R. Bartlett, U. A. Newton, R. A. White et N. L. Jones « Legionella in Cooling Water Systems » J. Hyg. Camb. 1982, 88, 369-381.
12. E. E. Sutherland et S. G. Berk « Survie des protozoaires dans les biocides de la tour de refroidissement » J. Ind. Microbiol. 1996, 16, 73-78.
13. D. B. McIlwaine, J. Diemer et S. Berk, « L’efficacité du glutaraldéhyde contre les protozoaires porteurs de la légionelle », Corrosion 98, article no 98521, (San Diego CA, NACE 1998)
14. Agence de protection de l’environnement. (s.d.). EPA : Système national d’élimination des rejets de polluants (NPDES). Le 22 novembre 2022, extrait du site https://www.epa.gov/npdes
15. Baron, C. D. « Novel, Oxydant doux, a amélioré le rendement du traitement de l’eau de refroidissement par rapport aux oxydants traditionnels. » Institut de technologie de refroidissement, mars 2012
16. Baron, C. D. « Améliorer la performance du système et réduire la corrosion du système à l’aide d’un nouveau biocide pour les tours de refroidissement. » Institut de technologie de refroidissement, février 2016
17. Note technique relative aux biocides Rohm et Haas, « Stoichiométrie du microbiocide KATHON WT à 1,5 % avec bisulfite de sodium ».
18. Merianos, J. J. 1991. « Composés antimicrobiens d’ammonium quaternaire » dans Block, S. S., Désinfection, stérilisation et préservation, 4e édition, Lea & Febiger, Philadelphie, p. 225 à 255, et les références qui y sont citées.
19. Petrocci, A. N., Green, H. A., Merianos, J. J. et Like, B., 1974. « The Properties of Dialkyl Dimethyl Quaternary Ammonium Compounds », C. S. M. A. Proceedings of the 60th Mid Year Meeting, mai 1974, p. 87 à 89.
20. R. M. Post, K. Emery, G. Dombrowski et M. Fagan, « Nettoyage efficace du remplissage de la tour de refroidissement en plastique cellulaire », du 33e atelier annuel sur la chimie des services publics d’électricité, 11 juin-13, 2013, Champaign, Illinois.
21. K. Boudreaux, « Using Biodetergents to Recover Megawatts Cheaply and with Minimal Environmental Impact », Power Plant Chemistry, 2015, vol. 17(3), p. 168 à 177.
22. R. D. Vidic. S. M. Duda et J. E. Stout, « Contrôle biologique des systèmes d’eau de refroidissement utilisant des dispositifs de traitement non chimiques », numéro de projet ASHRAE 1361-RP, rapport technique final, avril 2010.
23. J. E. Stout, S. M. Duda et R. D. Vidic, « Efficacité des dispositifs non chimiques dans le contrôle de la légionelle : Résultats d’un modèle de système de refroidissement » Institut de technologie de refroidissement Papier n° : TP11-08.
24. Khalanski, M. (1993). Essai de cinq méthodes pour le contrôle des moules zébrées dans les circuits de refroidissement des centrales électriques situées sur la rivière Moselle. Troisième Conférence internationale de moules zébrées, Toronto.
25. Matisoff, G., Brooks, G. et Bourland, I.B. (1996). Optimiser les processus de traitement du dioxyde de chlore aux moules pour adultes. J. A.W.W.A., août 1996, p. 93-106.
26. Roensch, F., « Contrôle des palourdes et des moules » Bulletin technique ChemTreat n° 91, septembre 2004.
27. Elliott, P., « Recommandations de prétraitement, de passivation et d’entretien pour les tours de refroidissement galvanisées ou les condenseurs à évaporation », Bulletin technique ChemTreat n° 82, avril 2021.
À propos des auteurs

Pete Elliott
Conseiller principal en personnel technique
Pete Elliott est un expert technique de confiance possédant plus de 30 ans d’expérience dans les solutions de traitement de l’eau pour les systèmes de traitement des services publics et thermique. Dans son domaine de travail, Pete se concentre sur la production de pratiques exemplaires pour des opérations d’usine efficaces, la conservation de l’énergie et de l’eau, ainsi que la protection et la préservation des actifs. Il a publié des articles techniques pour le Cooling Technologies Institute (CTI) et l’Association of Water Technologies (AWT). Pete est titulaire d’un baccalauréat en génie civil de l’Université Villanova et a occupé le poste d’agent d’ingénierie dans la Marine américaine.

Rajendra P. Kalakodimi, Ph. D.
Directeur de l’eau de refroidissement
Rajendra P. Kalakodimi est directeur de l’eau de refroidissement chez ChemTreat. Il a obtenu une maîtrise en chimie physique à l’Université Andhra et un doctorat en électrochimie à l’Indian Institute of Science de Bangalore en 2003. En Inde, le Dr Kalakodimi a occupé le poste de chef technique de l’ingénierie au Centre de technologie de GE Inde à Bangalore et de directeur de produit pour les produits chimiques et les solutions de surveillance pour GE Water. Il a plus de 20 dépôts de brevets, 20 publications internationales et diverses présentations de conférences.

Richard H. Tribble
Consultant en services techniques
Richard H. Tribble est diplômé de la Virginia Commonwealth University et travaille dans le traitement de l’eau depuis 26 ans. Il a développé une multitude de formulations de produits de refroidissement haute performance, possède 6 brevets sous son nom et a rédigé de nombreux articles. Il travaille actuellement comme champion des produits en boucle fermée pour ChemTreat et se spécialise dans l’analyse des causes profondes des défaillances.
Remerciements
Le manuel des essentiels de l’eau ChemTreat n’aurait pas été possible sans les contributions de nombreuses personnes. Voir la liste complète des contributeurs.
